


Ataxiile spinocerebeloase (SCA) reprezintă un grup complex de peste 40 de afecțiuni neurologice ereditare, caracterizate prin degenerarea progresivă a cerebelului și a măduvei spinării. Aceste boli afectează coordonarea, echilibrul, vorbirea și mișcările, având un impact semnificativ asupra calității vieții. Deși nu există un leac, diverse terapii și strategii de management pot ameliora simptomele și pot încetini progresia bolii, oferind pacienților o speranță de viață normală sau doar ușor redusă în multe cazuri.
- 🧬 Ce sunt? Un grup de peste 40 de boli genetice neurodegenerative care afectează coordonarea motorie.
- 🌍 Cât de rare sunt? Prevalența globală este de 1-5 cazuri la 100.000 de persoane, variind în funcție de regiune și tipul specific de SCA.
- 🩺 Simptome principale: Probleme de echilibru și mers (ataxie), vorbire neclară (dizartrie), dificultăți de înghițire (disfagie) și mișcări oculare anormale (nistagmus).
- ⏳ Cum progresează? Evoluția este lentă, de obicei pe parcursul a 10-20 de ani, ducând la necesitatea utilizării unui scaun cu rotile.
- 💊 Există tratament? Nu există un tratament curativ, dar managementul simptomatic prin fizioterapie, logopedie și medicamente poate îmbunătăți semnificativ calitatea vieții.
Cuprins
🧬 Introducere: Ce sunt ataxiile spinocerebeloase (SCAs)?
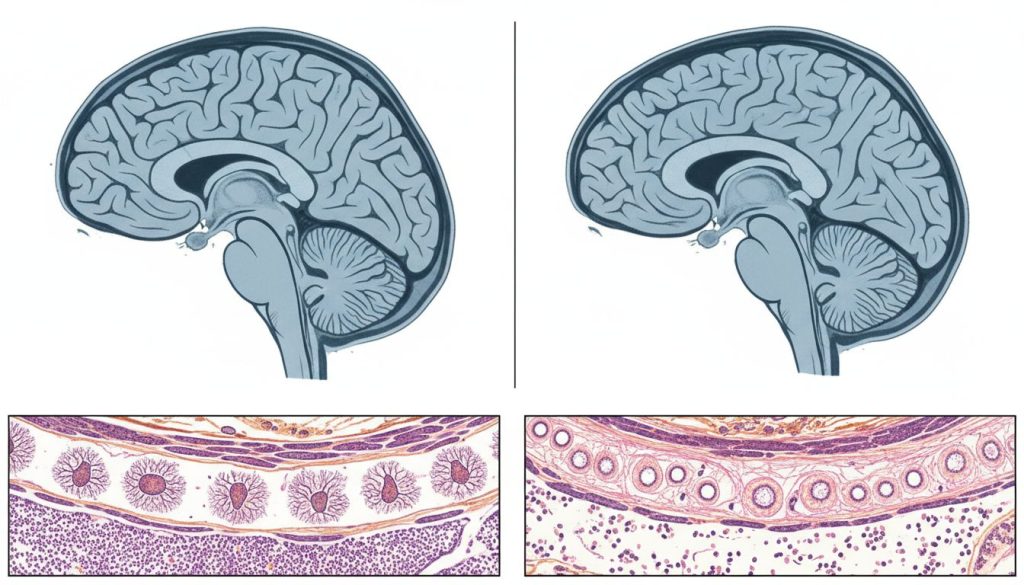
Ataxiile spinocerebeloase (SCAs) reprezintă un grup eterogen și complex de tulburări neurodegenerative, ereditare, care afectează în principal cerebelul – centrul de comandă al creierului pentru coordonarea mișcărilor. Termenul “ataxie” provine din greacă și înseamnă “lipsă de ordine”, descriind perfect dificultatea pacienților de a controla mișcările voluntare. Progresia acestor boli duce la deteriorarea treptată a coordonării (ataxie), echilibrului, vorbirii și, în unele cazuri, a altor funcții neurologice.
Fiecare tip de SCA este numerotat în ordinea descoperirii genei cauzatoare (de la SCA1 la SCA48 și mai departe). Deși toate au ca numitor comun ataxia, simptomele specifice, vârsta de debut și rata de progresie variază considerabil între tipuri și chiar între membrii aceleiași familii.
Tipuri genetice identificate
Prevalență globală
Risc de transmitere la copii
Cel mai frecvent tip global
Debutul simptomelor are loc, în general, la vârsta adultă, între 20 și 50 de ani, dar există și forme juvenile sau cu debut tardiv. Din păcate, speranța de viață poate fi redusă în formele mai agresive, decesul survenind adesea din cauza complicațiilor respiratorii sau cardiace, nu direct din cauza ataxiei.
🩺 Simptome comune și complicații
Manifestările clinice ale SCA sunt dominate de disfuncția cerebeloasă, dar pot include și o gamă largă de alte semne neurologice, reflectând afectarea altor părți ale sistemului nervos.
Simptome comune
Simptomele de bază sunt întâlnite în majoritatea tipurilor de SCA, deși severitatea și ordinea apariției lor pot varia:
- Ataxia de mers și echilibru: Este adesea primul și cel mai evident simptom. Persoanele afectate au un mers instabil, cu picioarele depărtate, similar cu cel al unei persoane în stare de ebrietate. Căderile devin frecvente și imprevizibile.
- Dismetria membrelor: Reprezintă incapacitatea de a controla distanța, viteza și forța unei mișcări. Se manifestă prin dificultăți la activități de finețe, cum ar fi scrisul de mână, încheierea nasturilor sau atingerea unui obiect cu precizie (de exemplu, dusul unui pahar la gură).
- Dizartria: Vorbirea devine lentă, neclară, sacadată și cu un ritm anormal. Pacienților le este greu să articuleze cuvintele, ceea ce face comunicarea dificilă.
- Nistagmusul și alte tulburări oculare: Mișcări involuntare, rapide și ritmice ale ochilor. Pot apărea și dificultăți în urmărirea obiectelor cu privirea sau vedere dublă (diplopie).
Progresia tipică a simptomelor
În general, simptomele debutează subtil și se agravează lent. Progresia medie duce la dependența de un scaun cu rotile în 10-20 de ani de la debut, în special în tipuri precum SCA1 și SCA2. Managementul proactiv poate încetini această evoluție.
Simptome rare și complicații
Pe măsură ce boala avansează și afectează și alte zone ale creierului și sistemului nervos, pot apărea simptome suplimentare, unele specifice anumitor tipuri de SCA:
- Disfagie: Dificultăți la înghițire, inițial pentru lichide, apoi și pentru solide. Aceasta crește riscul de înec și de pneumonie de aspirație (infecție pulmonară cauzată de pătrunderea alimentelor în căile respiratorii).
- Neuropatie periferică: Afectarea nervilor de la nivelul membrelor, cauzând amorțeli, furnicături, durere sau slăbiciune musculară. Este frecventă în SCA3 și SCA4.
- Tulburări de mișcare extrapiramidale: Pot include distonie (contracții musculare involuntare), coree (mișcări bruște, necontrolate) sau semne de parkinsonism (rigiditate, tremor de repaus, mișcări lente).
- Afectare cognitivă: Unele tipuri de SCA pot fi asociate cu dificultăți de memorie, planificare și alte funcții executive.
- Cardiomiopatie: Slăbirea mușchiului inimii, o complicație serioasă observată în special în SCA8.
- Complicații musculo-scheletice: Pe termen lung, imobilitatea și posturile anormale pot duce la scolioză, contracturi articulare și osteoporoză.
🔬 Tipuri principale de SCAs
Deși au fost identificate peste 40 de tipuri, câteva dintre ele sunt mult mai frecvente și mai bine studiate. Tabelul de mai jos prezintă o comparație între cele mai comune tipuri de ataxie spinocerebeloasă.
Distribuția tipurilor de SCA
| Tip SCA | Prevalență globală | Vârstă medie de debut | Simptome cheie distinctive | Prognostic și rata de progresie |
|---|---|---|---|---|
| SCA1 | 2-6% | 30-40 ani | Ataxie severă, dizartrie, semne piramidale (reflexe exagerate, spasticitate). | Progresie rapidă. Speranța de viață este în medie de 15 ani de la debut. |
| SCA2 | 10-15% | 20-30 ani | Ataxie cu progresie lentă, mișcări oculare foarte lente (sacade), tremor, hipotonie. | Progresie rapidă, dar variabilă. Poate avea debut juvenil. |
| SCA3 (Boala Machado-Joseph) | ~50% | 30-40 ani | Ataxie, distonie, oftalmoplegie (paralizia mușchilor oculari), neuropatie periferică, semne de parkinsonism. | Foarte variabil. Supraviețuirea poate depăși 20 de ani de la debut. |
| SCA6 | 10-15% | 50+ ani | Ataxie “pură”, adesea episodică la început, cu progresie foarte lentă. Vertij și nistagmus proeminente. | Progresie foarte lentă, speranță de viață adesea normală. |
| SCA7 | 5-10% | 20-40 ani | Ataxie însoțită de pierderea progresivă a vederii (retinopatie pigmentară), ducând la orbire. | Progresie rapidă, în special în cazurile cu debut timpuriu. |
🧬 Cauze și factori de risc
Ataxiile spinocerebeloase sunt afecțiuni pur genetice, cauzate de mutații într-o singură genă.
Această expansiune duce la producerea unei proteine anormale, alungite, care nu se poate plia corect. Aceste proteine toxice se acumulează în celulele nervoase, formând agregate care interferează cu funcționarea normală a neuronilor și, în cele din urmă, duc la moartea acestora. Acest proces de degenerare neuronală este cel care stă la baza atrofiei cerebeloase și a simptomelor clinice.
Factori de risc:
- Istoricul familial: Acesta este singurul factor de risc major. SCAs se transmit într-un mod dominant autosomal. Acest lucru înseamnă că o singură copie a genei mutante de la un părinte este suficientă pentru a dezvolta boala. Fiecare copil al unei persoane afectate are un risc de 50% de a moșteni gena și de a dezvolta boala.
- Anticipația genetică: În multe SCAs de tip CAG, expansiunea tinde să devină mai mare la fiecare generație. O expansiune mai mare este adesea asociată cu un debut mai timpuriu al bolii și cu o formă mai severă.
Nu există factori de risc legați de stilul de viață (dietă, fumat, etc.) care să cauzeze boala. Prevenirea genetică nu este posibilă odată ce mutația este prezentă, dar consilierea genetică joacă un rol crucial pentru familiile afectate.
🩺 Diagnostic
Diagnosticul de SCA se bazează pe o combinație de evaluare clinică, imagistică cerebrală și, cel mai important, testare genetică.
Metode de diagnostic
Procesul de diagnosticare este un parcurs în mai multe etape, care vizează atât confirmarea prezenței ataxiei, cât și excluderea altor cauze posibile.
-
Examen clinic și istoric familialMedicul neurolog va efectua un examen fizic detaliat pentru a evalua echilibrul, coordonarea, reflexele, forța musculară și vorbirea. O anamneză amănunțită a istoricului medical al familiei este esențială pentru a identifica un posibil model de moștenire.
-
Imagistică prin rezonanță magnetică (RMN)Un RMN cerebral este efectuat pentru a vizualiza structura creierului. În cazul SCA, RMN-ul poate dezvălui atrofie (micșorare) a cerebelului și, uneori, a trunchiului cerebral. De asemenea, ajută la excluderea altor cauze de ataxie, cum ar fi tumorile, accidentul vascular cerebral (AVC) sau scleroza multiplă.
-
Testare geneticăAcesta este pasul final și definitiv. O probă de sânge este analizată pentru a căuta expansiuni anormale ale repetițiilor CAG în genele asociate cu cele mai comune tipuri de SCA. De obicei, se folosește un panel de testare care acoperă mai multe tipuri de SCA simultan.
Când să consulți un medic
Este crucial să consulți un medic neurolog dacă prezinți oricare dintre următoarele situații:
- Observi o deteriorare progresivă a echilibrului sau a coordonării, fără o cauză evidentă.
- Ai membri în familie diagnosticați cu ataxie sau cu o tulburare neurologică neidentificată.
- Vorbirea ta a devenit neclară sau ai dificultăți de înghițire.
- Simptomele tale nu pot fi explicate prin alte condiții, cum ar fi consumul de alcool, medicamente sau alte boli cunoscute.
🧠 Verificarea cunoștințelor
Care este principalul factor de risc pentru dezvoltarea unei ataxii spinocerebeloase (SCA)?
💊 Tratament și management
În prezent, nu există un tratament care să vindece ataxiile spinocerebeloase sau să oprească complet progresia neurodegenerării. Cu toate acestea, există o abordare multidisciplinară care se concentrează pe managementul simptomelor, menținerea funcționalității și îmbunătățirea calității vieții.
Tratamente disponibile (simptomatice)
- Fizioterapie: Esențială pentru a menține mobilitatea, forța musculară și echilibrul pentru cât mai mult timp posibil. Poate îmbunătăți stabilitatea mersului cu 20-30%.
- Terapie ocupațională: Ajută pacienții să se adapteze la limitările fizice prin utilizarea de dispozitive ajutătoare (bastoane, cadre, scaune cu rotile) și prin modificarea mediului de acasă.
- Logopedie (terapia vorbirii): Îmbunătățește dizartria și oferă strategii pentru a face față disfagiei, reducând riscul de aspirație.
- Medicamente: Pot fi folosite pentru a controla simptome precum rigiditatea, spasticitatea, depresia sau tulburările de somn. Riluzolul este studiat în trialuri clinice pentru efectul său neuroprotector.
Limitări și provocări
- Nu există un leac sau un tratament care să oprească moartea neuronală.
- Terapiile genice sunt încă în faze incipiente de cercetare (Faza I) și nu sunt disponibile pe scară largă.
- Managementul necesită o echipă medicală complexă și un angajament pe termen lung din partea pacientului și a familiei.
- Costurile asociate cu terapiile, adaptările locuinței și îngrijirea pot fi substanțiale.
Managementul este adaptat în funcție de stadiul bolii și de simptomele specifice fiecărui pacient. De exemplu, pentru pacienții cu SCA3, monitorizarea cardiacă este importantă, în timp ce pentru cei cu SCA6, medicamente precum acetazolamida pot reduce frecvența episoadelor de ataxie. Diferențele de tratament bazate pe sex sunt inexistente, deoarece boala afectează ambele sexe în mod egal. Totuși, abordarea terapeutică variază cu vârsta: copiii cu debut precoce (ex. SCA2) necesită terapie intensivă și suport educațional, în timp ce la vârstnicii cu forme cu progresie lentă (ex. SCA6), accentul se pune pe prevenirea căderilor.
🧘 Stil de viață și prevenție
Deși nu se poate preveni apariția bolii dacă mutația genetică este prezentă, un stil de viață adaptat poate ajuta la gestionarea simptomelor și la menținerea independenței.
- Exerciții fizice regulate: Activități precum înotul, ciclismul staționar și exercițiile de stretching ajută la menținerea tonusului muscular și a flexibilității.
- Alimentație echilibrată: O dietă sănătoasă ajută la menținerea unei greutăți optime, reducând presiunea asupra articulațiilor. Pacienții cu disfagie pot necesita o dietă cu textură modificată.
- Evitarea alcoolului: Alcoolul afectează cerebelul și poate agrava semnificativ simptomele de ataxie.
- Consiliere genetică: Pentru familiile afectate, consilierea genetică este esențială. Aceasta oferă informații despre riscul de transmitere și prezintă opțiuni reproductive, cum ar fi diagnosticul genetic preimplantare (PGD), care permite selectarea embrionilor neafectați în cadrul fertilizării in vitro.
Adaptarea locuinței
Modificări simple, cum ar fi instalarea de bare de sprijin în baie, îndepărtarea covoarelor care pot cauza împiedicare și asigurarea unei bune iluminări, pot reduce semnificativ riscul de căderi și pot crește siguranța pacientului acasă.
❓ Întrebări frecvente (FAQ)
▼
Rata de progresie variază foarte mult în funcție de tipul de SCA și de numărul de repetiții CAG. În general, evoluția este lentă, desfășurându-se pe parcursul a 10-20 de ani până la stadiul în care este necesar un scaun cu rotile. Tipurile precum SCA1 progresează mai rapid, în timp ce SCA6 are o evoluție mult mai lentă.
▼
SCAs se moștenesc într-un mod dominant autosomal. Acest lucru înseamnă că dacă un părinte are boala, fiecare copil are un risc de 50% de a moșteni gena mutantă și de a dezvolta boala, indiferent de sex. Persoanele care nu moștenesc gena nu vor dezvolta boala și nu o pot transmite mai departe.
▼
Speranța de viață depinde de tipul de SCA și de severitatea bolii. Pentru multe tipuri, cum ar fi SCA6 sau formele cu debut tardiv de SCA3, speranța de viață poate fi normală sau aproape normală. În formele mai agresive, cum ar fi SCA1 sau cele cu debut juvenil, speranța de viață poate fi redusă din cauza complicațiilor, în special cele respiratorii.
📚 Referințe / Surse
Surse medicale internaționale
Informațiile prezentate în acest articol sunt bazate pe cercetări și ghiduri de la instituții medicale de renume. Mai jos sunt câteva dintre sursele principale consultate:
- National Center for Biotechnology Information (NCBI). (n.d.). Hereditary Ataxia Overview – GeneReviews®. www.ncbi.nlm.nih.gov/books/NBK1138/
- Mayo Clinic. (n.d.). Ataxia – Symptoms and causes. www.mayoclinic.org/diseases-conditions/ataxia/symptoms-causes/syc-20355652
- National Health Service (NHS). (n.d.). Ataxia – Types. www.nhs.uk/conditions/ataxia/symptoms/
- Cleveland Clinic. (n.d.). Ataxia: What It Is, Causes, Symptoms, Treatment & Types. my.clevelandclinic.org/health/symptoms/17748-ataxia
- National Ataxia Foundation. (n.d.). Episodic Ataxia. www.ataxia.org/ea/




