


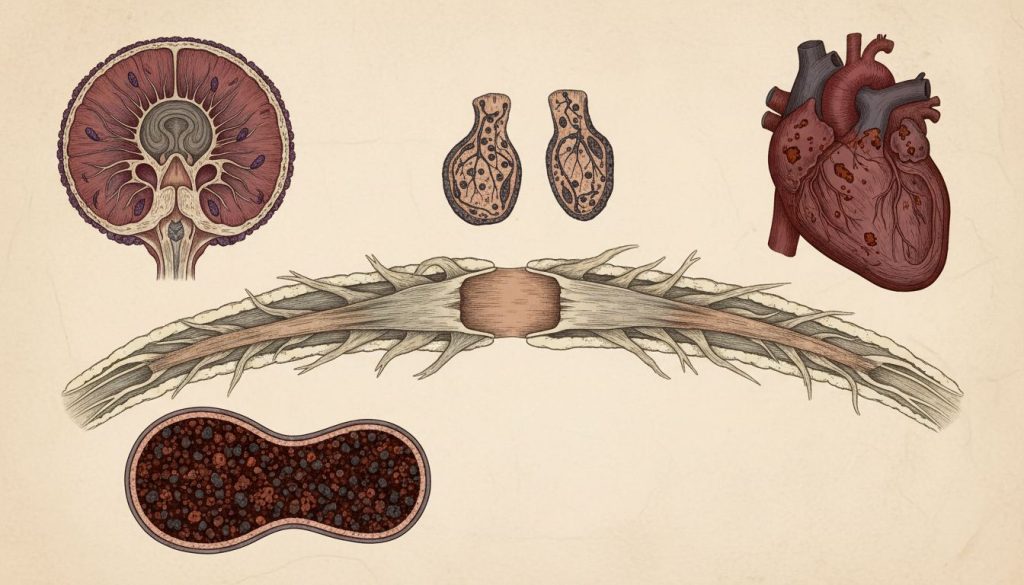
Ataxia Friedreich (FRDA) este cea mai frecventă formă de ataxie ereditară, o afecțiune neurodegenerativă rară care se manifestă de obicei în copilărie sau adolescență. Cauzată de o mutație genetică pe cromozomul 9, boala duce la o producție deficitară a proteinei frataxină, esențială pentru funcționarea mitocondriilor. Acest deficit afectează progresiv sistemul nervos, inima și pancreasul. Pacienții se confruntă cu dificultăți de mers și coordonare, probleme de vorbire, deformări scheletice precum scolioza și, în multe cazuri, complicații cardiace grave (cardiomiopatie hipertrofică) și diabet. Deși nu există un tratament curativ, managementul multidisciplinar, care include fizioterapie, logopedie, monitorizare cardiacă și îngrijire ortopedică, este esențial pentru a îmbunătăți calitatea vieții și a gestiona simptomele.
- 🧬 Cauza genetică: Ataxia Friedreich este o boală autozomal recesivă cauzată de o mutație în gena FXN de pe cromozomul 9.
- 🚶 Simptome principale: Boala debutează de obicei între 10 și 15 ani cu dificultăți de mers (ataxie), pierderea coordonării, vorbire neclară (dizartrie) și slăbiciune musculară.
- ❤️ Complicații severe: Cardiomiopatia hipertrofică este cea mai frecventă complicație și principala cauză de deces, alături de riscul crescut de diabet zaharat.
- 🚫 Tratament: În prezent, nu există un tratament care să vindece boala, dar terapiile de susținere (fizioterapie, logopedie) și managementul complicațiilor pot ameliora simptomele și prelungi speranța de viață.
Cuprins
🧬 Introducere – Ce este Ataxia Friedreich?
Ataxia Friedreich (FRDA) este o boală neurodegenerativă rară, ereditară, care provoacă leziuni progresive ale sistemului nervos. Descrisă pentru prima dată în 1863 de medicul german Nikolaus Friedreich, aceasta este cea mai frecventă formă de ataxie ereditară, afectând aproximativ 1 din 20.000 până la 1 din 50.000 de persoane în populațiile europene. Termenul “ataxie” se referă la pierderea coordonării mișcărilor voluntare, iar în cazul FRDA, aceasta se manifestă prin dificultăți de mers, probleme de echilibru, afectarea vorbirii și slăbiciune musculară.
Boala afectează cerebelul, măduva spinării și nervii periferici, ducând la o degenerare treptată a acestor structuri. Pe lângă simptomele neurologice, Ataxia Friedreich are un impact sistemic, afectând adesea inima (provocând cardiomiopatie hipertrofică) și pancreasul (crescând riscul de diabet). Datorită naturii sale progresive și a complicațiilor multisistemice, FRDA reprezintă o provocare clinică semnificativă, necesitând o abordare terapeutică complexă și multidisciplinară pentru a menține calitatea vieții pacienților.
🔬 Genetică și Mecanisme Moleculare
La baza Ataxiei Friedreich stă o cauză pur genetică. Boala este moștenită într-un model autozomal recesiv, ceea ce înseamnă că o persoană trebuie să moștenească două copii ale genei defecte – una de la fiecare părinte – pentru a dezvolta afecțiunea. Părinții, având o singură copie a genei mutante, sunt purtători sănătoși și, de obicei, nu prezintă niciun simptom.
Riscul de moștenire
- 25% șanse de a moșteni două gene mutante și de a dezvolta boala.
- 50% șanse de a moșteni o singură genă mutantă și de a fi purtător sănătos.
- 25% șanse de a moșteni două gene normale.
Mutația genei FXN și proteina frataxină
Gena responsabilă pentru FRDA se numește FXN și este localizată pe cromozomul 9 (mai exact, în regiunea 9q13-q21.1). La persoanele sănătoase, această genă conține o secvență repetitivă a trei nucleotide – Guanină-Adenină-Adenină (GAA). În mod normal, această secvență se repetă de 5 până la 33 de ori.
La pacienții cu Ataxie Friedreich, această secvență GAA este extinsă anormal, repetându-se de la 66 până la peste 1.700 de ori. Majoritatea pacienților au între 600 și 1.200 de repetiții. Această expansiune masivă a tripletului GAA împiedică citirea corectă a genei FXN, ceea ce duce la o producție semnificativ redusă a proteinei numite frataxină.
Frataxina este o proteină esențială pentru funcționarea normală a mitocondriilor – “uzinele energetice” ale celulelor. Ea joacă un rol crucial în formarea clusterelor de fier-sulf, care sunt componente vitale ale multor enzime implicate în producerea de energie (respirația celulară) și în protecția împotriva stresului oxidativ. Nivelurile scăzute de frataxină duc la:
- Acumularea toxică de fier în mitocondrii.
- Creșterea stresului oxidativ și deteriorarea celulară.
- Producție deficitară de energie (ATP).
Celulele nervoase și cele musculare (inclusiv cele cardiace), care au un consum energetic foarte mare, sunt deosebit de vulnerabile la acest deficit, ceea ce explică manifestările neurologice și cardiace ale bolii.
🏃♂️ Simptome și Debut Clinic
Ataxia Friedreich este o boală progresivă, iar simptomele se agravează în timp. Vârsta de debut este un factor predictiv important pentru severitatea bolii.
-
Debut tipic (10-15 ani): Cele mai multe cazuri (aproximativ 75%) se încadrează în această categorie. Primele semne sunt de obicei subtile și includ dificultăți de echilibru, împiedicări frecvente și o neîndemânare generală.
-
Debut tardiv (după 25 de ani): Această formă, cunoscută ca Late Onset Friedreich’s Ataxia (LOFA), are o progresie mai lentă și simptome adesea mai blânde.
-
Debut foarte tardiv (după 40 de ani): Cunoscută ca Very Late Onset Friedreich’s Ataxia (VLOFA), această variantă este mult mai rară și are o evoluție lentă.
Simptomele principale progresive
Pe măsură ce boala avansează, simptomele devin mai pronunțate și mai diverse, afectând multiple sisteme ale organismului.
Simptome neurologice
- Ataxia de mers: Acesta este simptomul definitoriu și, de obicei, primul care apare. Mersul devine instabil, cu pași neregulați și o bază largă de susținere pentru a menține echilibrul.
- Dizartria: Vorbirea devine lentă, neclară și sacadată (“slurred speech”), din cauza lipsei de coordonare a mușchilor implicați în fonație.
- Ataxia membrelor: Coordonarea fină a mâinilor și picioarelor se deteriorează, făcând dificile activități precum scrisul, mâncatul sau încheierea nasturilor.
- Slăbiciune musculară: În special la nivelul picioarelor, contribuind și mai mult la dificultățile de mers.
- Pierderea reflexelor tendinoase: În special la nivelul genunchilor și gleznelor.
- Pierderea sensibilității: Pacienții își pierd treptat simțul poziției (propriocepția) și simțul vibratoriu, în special la nivelul extremităților.
Complicații cardiace
Afectarea cardiacă este extrem de frecventă și reprezintă principala cauză de morbiditate și mortalitate în Ataxia Friedreich.
Cardiomiopatia hipertrofică
Aproximativ două treimi dintre pacienți dezvoltă cardiomiopatie hipertrofică, o afecțiune în care mușchiul inimii (miocardul) se îngroașă anormal. Aceasta poate duce la insuficiență cardiacă, aritmii (bătăi neregulate ale inimii) și risc crescut de moarte subită.
Complicații scheletice și endocrine
- Scolioza: O curbură anormală a coloanei vertebrale apare la majoritatea pacienților și poate necesita corecție chirurgicală dacă este severă.
- Pes cavus: Deformarea piciorului, caracterizată printr-un arc plantar foarte înalt.
- Diabetul zaharat: Aproximativ 10-20% dintre pacienți dezvoltă diabet, iar mulți alții prezintă intoleranță la glucoză, din cauza afectării celulelor pancreatice producătoare de insulină.
🩺 Diagnostic
Diagnosticul Ataxiei Friedreich se bazează pe o combinație de evaluare clinică, istoric familial și teste de confirmare. Suspiciunea apare la un copil sau adolescent care dezvoltă progresiv ataxie de mers, dizartrie și pierderea reflexelor.
-
Evaluare clinică neurologicăMedicul neurolog evaluează mersul, coordonarea, vorbirea, reflexele, forța musculară și sensibilitatea. Prezența semnelor caracteristice ridică suspiciunea de FRDA.
-
Testare geneticăDiagnosticul este confirmat printr-un test de sânge care analizează gena FXN. Acest test genetic molecular măsoară numărul de repetiții ale tripletului GAA, fiind standardul de aur pentru diagnostic. Un număr de peste 66 de repetiții pe ambele alele confirmă diagnosticul.
-
Investigații suplimentareDupă confirmare, se efectuează alte investigații pentru a evalua amploarea afectării:
- Imagistică prin rezonanță magnetică (IRM): IRM-ul cerebral poate fi normal în stadiile incipiente, dar în stadii avansate poate arăta atrofie cerebeloasă. IRM-ul spinal este mai specific, relevând adesea atrofia măduvei spinării cervicale.
- Evaluare cardiologică: Electrocardiograma (ECG) și ecocardiografia sunt esențiale pentru a detecta și monitoriza cardiomiopatia.
- Teste de conducere nervoasă (EMG/NCV): Acestea arată de obicei o neuropatie senzorială axonală.
🧠 Verificare cunoștințe
Care este testul definitiv pentru confirmarea diagnosticului de Ataxie Friedreich?
🗂️ Tipuri și Variante
Deși majoritatea cazurilor urmează un model clasic, Ataxia Friedreich poate avea prezentări clinice diferite, în principal în funcție de vârsta de debut și de numărul de repetiții GAA.
Forma Clasică (Debut < 25 ani)
- Vârsta de debut este de obicei între 10 și 15 ani.
- Progresie mai rapidă a simptomelor neurologice.
- Risc ridicat și precoce de cardiomiopatie și scolioză.
- Pacienții ajung să necesite scaun rulant în aproximativ 10-15 ani de la debut.
- Asociată cu expansiuni GAA mai mari (de obicei > 300).
Forma cu Debut Tardiv (LOFA/VLOFA, > 25 ani)
- Simptomele apar după vârsta de 25 de ani, uneori chiar și după 40.
- Progresie mult mai lentă a bolii.
- Cardiomiopatia și scolioza sunt mai puțin frecvente sau mai puțin severe.
- Reflexele pot fi păstrate pentru o perioadă mai lungă.
- Asociată cu expansiuni GAA mai mici (de obicei < 300).
Există și variante genetice foarte rare, cum ar fi pacienții cu mutații punctiforme în gena FXN, care pot avea fenotipuri atipice. Corelația genotip-fenotip este puternică: în general, un număr mai mic de repetiții GAA se corelează cu un debut mai tardiv și o evoluție mai lentă a bolii, în timp ce expansiunile foarte mari sunt asociate cu forma clasică, severă.
⏳ Prognoză și Evoluție
Ataxia Friedreich este o afecțiune inexorabil progresivă, cu o deteriorare constantă a funcțiilor neurologice. Evoluția naturală a bolii duce la dizabilitate semnificativă pe parcursul a 10-20 de ani de la diagnostic.
Stadii Incipiente (Primii 5-10 ani)
• Mers încă posibil, dar instabil (ataxic)
• Neîndemânare și dificultăți de coordonare
• Vorbire ușor afectată
• Debutul scoliozei
Stadii Avansate (După 10-20 de ani)
• Pierderea capacității de a merge (necesită scaun rulant)
• Dizartrie severă, dificultăți de comunicare
• Slăbiciune musculară marcată
• Complicații cardiace și diabet
Severitatea și viteza progresiei variază considerabil de la un pacient la altul. Factorii principali care influențează prognosticul sunt:
- Vârsta de debut: Un debut mai timpuriu este asociat cu o progresie mai rapidă.
- Mărimea expansiunii GAA: Un număr mai mare de repetiții se corelează cu o boală mai severă.
- Prezența și severitatea cardiomiopatiei: Complicațiile cardiace sunt principalul factor care limitează speranța de viață.
Speranța de viață este, în medie, redusă, majoritatea pacienților trăind până în jurul vârstei de 40-50 de ani. Moartea survine cel mai adesea din cauza insuficienței cardiace sau a aritmiilor maligne.
❤️ Management și Tratament
În prezent, nu există un tratament curativ pentru Ataxia Friedreich care să oprească sau să inverseze progresia bolii. Cu toate acestea, un management suportiv și multidisciplinar este crucial pentru a ameliora simptomele, a preveni complicațiile și a îmbunătăți calitatea vieții.
Abordarea multidisciplinară
Îngrijirea optimă a unui pacient cu FRDA implică o echipă de specialiști, inclusiv: neurolog, cardiolog, endocrinolog, ortoped, fizioterapeut, logoped, terapeut ocupațional și consilier genetic.
Strategii de management
| Domeniu | Intervenții |
|---|---|
| Neurologic |
|
| Cardiac |
|
| Ortopedic |
|
| Metabolic |
|
Cercetarea medicală este foarte activă în acest domeniu, explorând diverse strategii terapeutice, cum ar fi terapia genică (pentru a înlocui gena FXN defectă), molecule care cresc expresia frataxinei și medicamente care combat efectele deficitului de frataxină (ex. antioxidanți).
🤔 Răspunsuri la Întrebări Cheie
▼
Nu, în prezent nu există un tratament curativ pentru Ataxia Friedreich. Toate intervențiile actuale sunt de susținere și vizează gestionarea simptomelor și a complicațiilor pentru a îmbunătăți calitatea vieții și a prelungi supraviețuirea. Există însă numeroase studii clinice în desfășurare care testează noi terapii promițătoare.
▼
Deoarece este o boală genetică, Ataxia Friedreich nu poate fi prevenită. Cuplurile cu istoric familial de FRDA sau care știu că sunt purtători pot beneficia de consiliere genetică. Opțiunile includ testarea prenatală sau diagnosticul genetic preimplantator (PGD) în cazul fertilizării in vitro, pentru a evalua riscul de a avea un copil afectat.
▼
Speranța de viață este variabilă și depinde în mare măsură de severitatea bolii, în special de gradul de afectare cardiacă. În medie, speranța de viață este de aproximativ 40-50 de ani. Cardiomiopatia hipertrofică și complicațiile sale (insuficiența cardiacă, aritmiile) reprezintă principala cauză de deces prematur.
▼
Majoritatea pacienților cu forma clasică a bolii vor necesita un scaun rulant pentru mobilitate la aproximativ 10-20 de ani de la debutul simptomelor. Pacienții cu forme cu debut tardiv (LOFA/VLOFA) pot să-și păstreze capacitatea de a merge pentru o perioadă mult mai lungă, unii chiar pentru toată viața.
📚 Resurse și Informații Suplimentare
Următoarele resurse oferă informații suplimentare despre Ataxia Friedreich, deși sunt disponibile în principal în limba engleză. Acestea pot fi utile pentru pacienți, familii și profesioniști din domeniul medical care doresc să aprofundeze cunoștințele despre această afecțiune.
- DocCheck Flexikon. Friedreich-Ataxie. flexikon.doccheck.com/de/Friedreich-Ataxie
- Ninja Nerd. (2020). What is Friedreich’s Ataxia? Causes, Symptoms & Inheritance. youtube.com/watch?v=ZbnROQNILPY
- Wikipedia. Friedreich-Ataxie. de.wikipedia.org/wiki/Friedreich-Ataxie




