


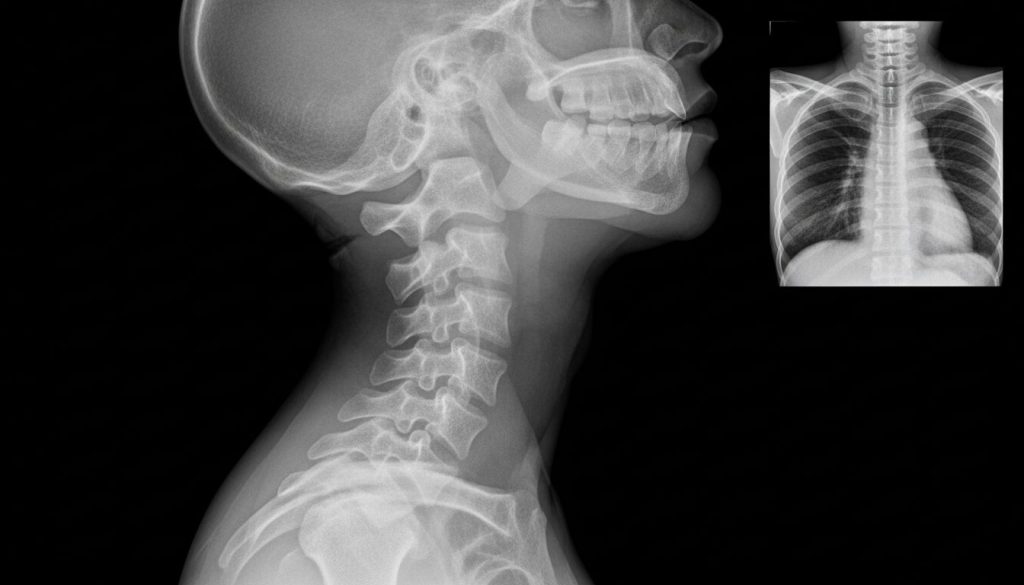
Sindromul Klippel-Feil (KFS) este o afecțiune congenitală rară, caracterizată prin fuziunea (unirea) a cel puțin două vertebre de la nivelul gâtului (coloana cervicală). Această anomalie structurală, care apare în timpul dezvoltării embrionare, poate duce la un gât vizibil mai scurt, mobilitate redusă și dureri cronice, dar manifestările variază semnificativ de la o persoană la alta. Deși triada clasică de simptome – gât scurt, linia joasă de inserție a părului pe ceafă și mișcare limitată a gâtului – a definit inițial sindromul, aceasta este prezentă la mai puțin de jumătate dintre pacienți.
Articolul de față explorează în detaliu cauzele, simptomele diverse, metodele de diagnostic și opțiunile de tratament pentru sindromul Klippel-Feil. Vom analiza clasificarea modernă a sindromului, anomaliile frecvent asociate (cardiace, renale, auditive) și impactul pe care îl are asupra calității vieții. Managementul se concentrează pe ameliorarea simptomelor prin terapie fizică și, în cazuri severe de instabilitate sau compresie nervoasă, prin intervenție chirurgicală. De asemenea, vom aborda recomandările privind stilul de viață, cum ar fi evitarea sporturilor de contact, pentru a preveni complicațiile neurologice grave.
- 🧬 Ce este? O fuziune congenitală a vertebrelor cervicale prezentă de la naștere.
- 📉 Cât de rar este? Afectează aproximativ 1 din 40.000 – 42.000 de nou-născuți.
- 🤔 Simptome cheie: Triada clasică (gât scurt, linie joasă a părului, mobilitate redusă) este prezentă la mai puțin de 50% dintre pacienți. Mulți pot avea doar dureri de gât sau pot fi asimptomatici.
- 🩺 Diagnostic: Se bazează pe examinări imagistice, precum radiografii, CT și RMN.
- 🤸♂️ Management: Majoritatea cazurilor sunt gestionate conservator (fizioterapie). Chirurgia este rezervată pentru instabilitate sau compresie neurologică.
- ⚠️ Precauții: Persoanele cu KFS trebuie să evite sporturile de contact și activitățile cu risc de traumatism la nivelul gâtului.
Cuprins
📜 Introducere: Ce este Sindromul Klippel-Feil?
Sindromul Klippel-Feil (KFS) este o anomalie congenitală rară a oaselor, definită prin fuziunea (sudarea) a două sau mai multe dintre cele șapte vertebre cervicale (vertebrele gâtului). Această afecțiune a fost descrisă pentru prima dată în 1912 de medicii francezi Maurice Klippel și André Feil, care au observat la pacienții lor o triadă clasică de caracteristici: un gât scurt, o linie de inserție a părului pe ceafă neobișnuit de joasă și o mobilitate redusă a capului și gâtului.
Deși această triadă este emblematică, studiile ulterioare au demonstrat că mai puțin de 50% dintre persoanele cu KFS prezintă toate cele trei caracteristici simultan. Spectrul sindromului este foarte larg: unii indivizi pot fi complet asimptomatici și diagnosticați întâmplător în urma unor investigații imagistice efectuate din alte motive, în timp ce alții pot prezenta deformări severe și complicații neurologice sau ale altor organe.
Frecvent, fuziunea apare la nivelul vertebrelor C2-C3. Prezența fuziunilor poate suprasolicita segmentele mobile adiacente ale coloanei vertebrale, accelerând uzura acestora (spondiloză) și crescând riscul de instabilitate, hernie de disc sau stenoză spinală. Din acest motiv, managementul corect și monitorizarea pe termen lung sunt esențiale pentru menținerea calității vieții.
🌡️ Simptome și manifestări clinice
Manifestările clinice ale sindromului Klippel-Feil sunt extrem de variabile, depinzând de numărul și localizarea vertebrelor fuzionate, precum și de prezența anomaliilor asociate.
Simptome comune
Simptomele direct legate de fuziunea vertebrală pot include:
- Durere cronică de gât (cervicalgie) și cefalee (dureri de cap): Acestea sunt cele mai frecvente acuze, adesea exacerbate de anumite mișcări.
- Rigiditate cervicală și mobilitate limitată: Dificultatea de a roti, apleca sau extinde gâtul este un semn distinctiv. Cu toate acestea, unii pacienți pot avea o mobilitate surprinzător de bună, compensată de segmentele vertebrale neafectate.
- Torticolis congenital: O postură anormală a capului, înclinat spre o parte și rotit spre partea opusă. Deși torticolisul poate avea multe cauze, inclusiv contracturi musculare (așa cum este descris în torticolisul muscular congenital), în contextul KFS, este de natură osoasă (torticolis osos).
- Asimetrie facială: Poate apărea ca o consecință a posturii anormale a capului menținute pe termen lung.
- Sunete (cracmente) la mișcarea gâtului.
Anomalii asociate frecvent
O caracteristică importantă a KFS este asocierea sa frecventă cu alte anomalii congenitale. Evaluarea unui pacient cu KFS trebuie să includă screening pentru aceste afecțiuni.
Scolioză (curbură laterală a coloanei)
Anomalii genito-urinare (ex: rinichi unic)
Probleme auditive (surditate)
Anomalii cardiace congenitale
- Anomalii ale coloanei vertebrale: Cea mai frecventă este scolioza congenitală (aproximativ 60% din cazuri). Pot apărea și alte deformări, cum ar fi cifoza.
- Malformația Sprengel: O afecțiune în care unul dintre omoplați este situat mai sus pe spate decât în mod normal (prezentă la 20-30% din pacienți).
- Anomalii genito-urinare: Afectează până la 30-50% dintre pacienți și pot include agenezia renală (absența unui rinichi), rinichi în potcoavă sau alte probleme structurale ale tractului urinar.
- Probleme auditive: Aproximativ 30% dintre pacienți pot avea un anumit grad de pierdere a auzului.
- Anomalii cardiovasculare: Se întâlnesc la 10-15% din cazuri și pot include defecte septale ventriculare.
- Simptome neurologice: Acestea sunt mai rare, dar pot fi severe. Pot include slăbiciune sau amorțeală în brațe (radiculopatie), probleme de echilibru și coordonare sau slăbiciune în picioare (mielopatie) dacă există compresie pe măduva spinării sau pe rădăcinile nervoase. Anomalia Kimmerle, o variantă anatomică a primei vertebre cervicale care poate comprima artera vertebrală, poate fi și ea prezentă și poate cauza simptome precum amețeli sau dureri de cap.
- Sincinezie: Mișcări involuntare în oglindă (de ex., mișcarea involuntară a unei mâini atunci când cealaltă mână se mișcă intenționat).
🧬 Cauze și factori de risc
Sindromul Klippel-Feil este rezultatul unui eșec în procesul normal de segmentare a somitelor cervicale, care are loc între a treia și a opta săptămână de dezvoltare embrionară. În mod normal, coloana vertebrală se dezvoltă din blocuri de țesut numite somite, care se separă pentru a forma vertebre individuale. În KFS, această separare nu se produce corect, ducând la fuziunea oaselor.
Factori genetici
- În majoritatea cazurilor, KFS apare sporadic, fără un istoric familial al bolii.
- Au fost identificate mutații în anumite gene, cum ar fi GDF6, GDF3 și MEOX1, care joacă un rol în dezvoltarea scheletului. Aceste mutații sunt însă rare.
- Există forme familiale, foarte rare, care pot fi moștenite autosomal dominant (o singură copie a genei mutante este suficientă) sau autosomal recesiv (sunt necesare două copii ale genei mutante).
Factori de risc non-genetici
- Etiologia exactă este în mare parte necunoscută.
- Nu au fost identificați factori de mediu sau maternali clari care să cauzeze KFS.
- Principalul “factor de risc” nu este pentru dezvoltarea sindromului, ci pentru apariția complicațiilor: persoanele cu KFS au un risc crescut de leziuni neurologice severe în urma unor traumatisme minore (căderi, accidente rutiere, activități sportive).
Atenție la traumatisme!
Hipermobilitatea segmentelor cervicale adiacente fuziunii face ca gâtul să fie vulnerabil la leziuni. O mișcare bruscă sau un impact care ar fi minor pentru o persoană fără KFS poate provoca fracturi, dislocări sau leziuni medulare catastrofale, inclusiv paralizie sau moarte subită. De aceea, restricționarea activităților cu risc este crucială.
📊 Tipuri de Sindrom Klippel-Feil (Clasificarea Samartzis)
Pentru a standardiza descrierea și a anticipa riscurile, a fost propusă o clasificare modernă de către Samartzis și colegii săi, care se bazează pe localizarea și extinderea fuziunii vertebrale. Aceasta a înlocuit clasificările mai vechi și este mai relevantă din punct de vedere clinic.
| Tip | Descriere | Prevalență | Caracteristici și riscuri tipice |
|---|---|---|---|
| Tip I | Fuziune congenitală a unui singur segment cervical (bloc vertebral). | ~25% | Adesea asimptomatic sau cu durere axială (localizată la nivelul gâtului). Risc mai mic de instabilitate, dar cu potențial de degenerare accelerată a discurilor adiacente. |
| Tip II | Multiple fuziuni non-contigue (segmente fuzionate separate de cel puțin o vertebră normală). | ~50% (cel mai frecvent) | Risc crescut de instabilitate la nivelul segmentelor mobile intercalate. Pacienții pot dezvolta spondiloză simptomatică și stenoză la o vârstă mai tânără. |
| Tip III | Multiple fuziuni contigue (mai multe vertebre fuzionate într-un singur bloc lung). | ~25% | Asociat cu cea mai severă deformare (gât foarte scurt și rigid). Risc înalt de instabilitate la joncțiunile cranio-cervicală și cervico-toracică, și risc crescut de compresie medulară. |
🩺 Diagnosticarea sindromului
Diagnosticul sindromului Klippel-Feil începe cu o suspiciune clinică bazată pe aspectul fizic și simptome, dar confirmarea se face prin metode imagistice.
-
Examinare fizicăMedicul evaluează aspectul gâtului, linia părului, simetria feței și, cel mai important, gama de mișcare a gâtului. Se efectuează și un examen neurologic complet pentru a depista eventuale deficite.
-
Radiografii (X-ray)Sunt esențiale pentru diagnostic. Radiografiile laterale ale coloanei cervicale arată clar fuziunea corpurilor vertebrale, a arcurilor posterioare și a proceselor spinoase. Radiografiile în flexie și extensie sunt cruciale pentru a evalua instabilitatea segmentelor mobile.
-
CT și RMNTomografia computerizată (CT) oferă o vizualizare superioară a anatomiei osoase și este utilă pentru planificarea chirurgicală. Rezonanța magnetică nucleară (RMN) este indispensabilă pentru a vizualiza țesuturile moi: măduva spinării, rădăcinile nervoase, discurile intervertebrale și ligamentele, fiind metoda de elecție pentru a detecta compresiile neurologice.
-
Investigații suplimentareDin cauza anomaliilor asociate frecvent, protocolul de diagnostic poate include o ecografie renală pentru a verifica rinichii și o evaluare cardiologică (EKG, ecocardiogramă) și audiologică.
🚨 Când să consulți un medic
Este esențial să solicitați o evaluare medicală specializată (ortopedie pediatrică, neurochirurgie sau genetică medicală) în următoarele situații:
Semne de alarmă care necesită un consult imediat:
- Apariția sau agravarea durerii de gât sau a durerilor de cap.
- Rigiditate progresivă a gâtului.
- Apariția unor simptome neurologice noi: amorțeli, furnicături, slăbiciune în mâini sau picioare, pierderea dexterității, probleme de mers sau echilibru.
- După orice traumatism la nivelul capului sau gâtului, chiar dacă pare minor.
- La un copil, dacă se observă o poziție anormală a capului (torticollis) sau o limitare evidentă a mișcării.
💊 Tratament și management
Nu există un tratament care să “vindece” sindromul Klippel-Feil, deoarece fuziunea vertebrală este permanentă. Prin urmare, managementul se concentrează pe controlul simptomelor, prevenirea complicațiilor și menținerea funcționalității.
Tratament Conservator (Non-chirurgical)
Este prima linie de tratament pentru majoritatea pacienților, în special pentru cei cu simptome ușoare până la moderate.
- Fizioterapie și kinetoterapie: Programe personalizate pentru a întări musculatura gâtului și a spatelui, a îmbunătăți postura și a menține flexibilitatea.
- Medicamente: Analgezice (paracetamol) și antiinflamatoare nesteroidiene (AINS, ex: ibuprofen) pentru a controla durerea. Relaxantele musculare pot fi utile în caz de spasme.
- Modificarea activităților: Evitarea mișcărilor repetitive sau a posturilor care exacerbează durerea.
- Orteze cervicale (gulere): Utilizate pe termen scurt pentru a limita mișcarea în perioadele de durere acută sau după traumatisme.
- Injecții spinale: Injecțiile cu corticosteroizi pot fi considerate pentru a reduce inflamația în cazul radiculopatiei (compresia unei rădăcini nervoase).
Tratament Chirurgical
Intervenția chirurgicală este rezervată cazurilor cu simptome severe și progresive, care nu răspund la tratamentul conservator.
- Indicații principale:
- Instabilitate cervicală: Mișcare excesivă și anormală a vertebrelor, care pune în pericol măduva spinării.
- Compresie neurologică: Simptome progresive de mielopatie (compresie pe măduvă) sau radiculopatie severă (compresie pe nerv).
- Deformare progresivă și severă.
- Durere cronică, invalidantă, refractară la tratament.
- Procedura principală: Artrodeza (fuziunea spinală). Chirurgul stabilizează segmentul instabil prin fuzionarea chirurgicală a vertebrelor afectate, folosind grefe osoase și implanturi metalice (plăci, șuruburi).
Diferențe de abordare (vârstă și sex)
- La copii: Obiectivul principal este prevenirea progresiei deformării și protejarea măduvei spinării. Abordarea chirurgicală poate necesita tehnici complexe (combinate anterior și posterior) pentru a preveni “fenomenul crankshaft” (o rotire continuă a coloanei în jurul fuziunii în timpul creșterii).
- La adulți: Chirurgia vizează decompresia structurilor nervoase și stabilizarea segmentelor uzate și instabile.
- În funcție de sex: Deși sindromul are o incidență aproximativ egală la ambele sexe, unele studii sugerează că femeile ar putea avea un risc mai mare de a prezenta anomalii asociate, necesitând o monitorizare mai atentă a altor sisteme de organe.
⚠️ Complicații posibile
Structura anormală a coloanei cervicale în KFS predispune pacienții la o serie de complicații pe termen lung, în special la nivelul segmentelor mobile care sunt suprasolicitate.
Frecvența și impactul complicațiilor pe termen lung
Foarte frecventă
Frecventă
Frecventă
Risc crescut
Risc crescut
Alte complicații includ durerea cronică invalidantă și limitarea progresivă a funcționalității, care pot afecta semnificativ calitatea vieții și capacitatea de muncă.
🧠 Verifică-ți cunoștințele!
Care element face parte din triada clasică a sindromului Klippel-Feil, chiar dacă este întâlnit la mai puțin de 50% dintre pacienți?
❤️🩹 Stil de viață, prevenție și FAQ
Adaptarea stilului de viață este fundamentală pentru pacienții cu sindrom Klippel-Feil, având ca scop principal minimizarea riscului de leziuni.
Recomandări privind stilul de viață
- Evitarea sporturilor de contact: Activități precum fotbalul, rugby-ul, hocheiul, artele marțiale și boxul sunt strict contraindicate.
- Prudență în activități cu risc de cădere: Gimnastica, călăria, schiul, scufundările și utilizarea trambulinelor trebuie evitate sau practicate cu extremă precauție și numai după acordul medicului specialist.
- Ergonomia la locul de muncă și acasă: Adaptarea scaunului, a biroului și a monitorului pentru a menține o postură corectă a gâtului.
- Monitorizare medicală regulată: Controale anuale sau la intervale stabilite de medic, chiar și în absența simptomelor, pentru a depista precoce eventualele semne de instabilitate sau degenerare.
Prevenție
Deoarece KFS este o afecțiune congenitală, nu poate fi prevenită. Totuși, se pot lua măsuri pentru prevenirea complicațiilor. În cazurile rare în care există un istoric familial sau o mutație genetică cunoscută, consilierea genetică poate fi utilă pentru a evalua riscul de transmitere la urmași.
Întrebări frecvente
▼
Este o afecțiune rară, cu o incidență estimată la 1 din 40.000 – 42.000 de nașteri la nivel global. Multe cazuri ușoare pot rămâne nediagnosticate.
▼
Nu, fuziunea vertebrală este permanentă. Tratamentul nu vizează vindecarea, ci managementul simptomatic și prevenirea complicațiilor. Calitatea vieții poate fi însă foarte bună cu un management adecvat.
▼
În România, managementul sindromului Klippel-Feil este realizat de echipe multidisciplinare. Puteți apela la medici specialiști în neurochirurgie sau ortopedie-traumatologie (în special cu supraspecializare în chirurgia spinală sau ortopedie pediatrică). Centrele universitare mari, precum Spitalul Universitar de Urgență București (SUUB) sau alte spitale cu secții de neurochirurgie și ortopedie de top, dispun de expertiza necesară pentru evaluarea și tratamentul acestei patologii complexe.
▼
Da, majoritatea persoanelor cu KFS, în special cele cu fuziuni limitate și fără instabilitate, pot duce o viață normală și activă, cu anumite precauții. Cheia este diagnosticarea corectă, înțelegerea limitărilor și respectarea recomandărilor medicale pentru a evita riscurile.
📚 Referințe / Surse
Nattiv, A., et al. (2023). Klippel Feil Syndrome. In: StatPearls [Internet]. StatPearls Publishing. www.ncbi.nlm.nih.gov/books/NBK493157/
Pediatric Orthopaedic Society of North America (POSNA). (n.d.). Klippel Feil Syndrome. posna.org/physician-education/study-guide/klippel-feil-syndrome
Orphanet. (2020). Isolated Klippel-Feil syndrome. www.orpha.net/en/disease/detail/2345
Wikipedia. (2023). Klippel–Feil syndrome. en.wikipedia.org/wiki/Klippel–Feil_syndrome
Cleveland Clinic. (2022). Klippel-Feil Syndrome (KFS). my.clevelandclinic.org/health/diseases/23919-klippel-feil-syndrome-kfs
Orthobullets. (n.d.). Klippel-Feil Syndrome. www.orthobullets.com/spine/2051/klippel-feil-syndrome






