


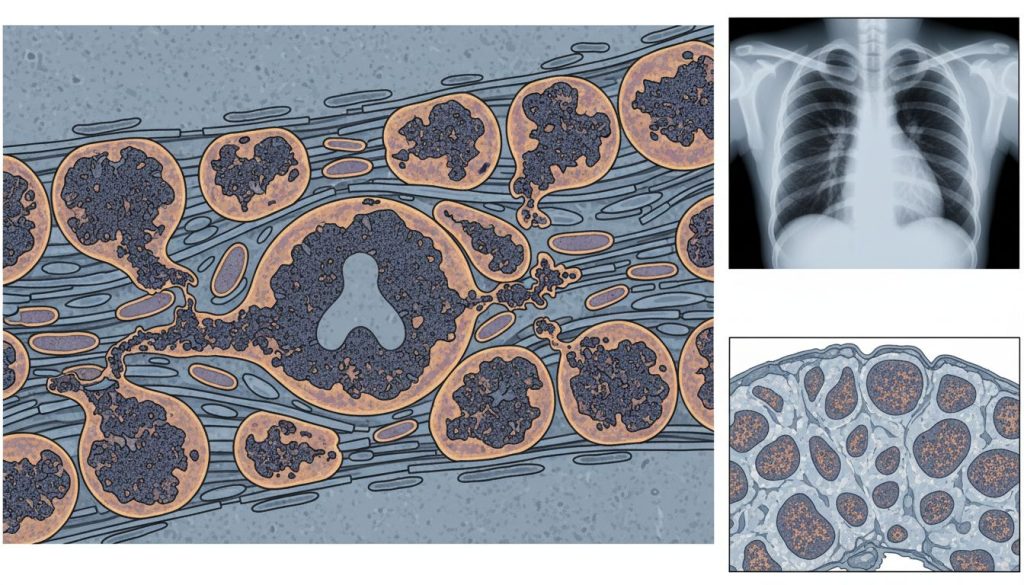
Boala Pompe, cunoscută și sub numele de glicogenoză de tip II, este o afecțiune genetică rară și progresivă, caracterizată printr-o afectare severă a musculaturii. Aceasta este cauzată de un deficit al enzimei alfa-glucozidaza acidă (GAA), ceea ce duce la acumularea de glicogen în lizozomii celulelor, în special în cele musculare. Fără această enzimă, celulele nu pot descompune glicogenul în glucoză pentru a produce energie, ceea ce slăbește progresiv mușchii, inclusiv cei responsabili pentru respirație și funcția cardiacă.
Articolul explorează în detaliu cele două forme principale ale bolii – forma infantilă, cu debut precoce și evoluție rapidă, și forma tardivă, care poate apărea oricând de la copilărie la maturitate. Vom discuta despre simptomele specifice, cauzele genetice, metodele de diagnostic precise și, cel mai important, opțiunile de tratament care au schimbat radical prognosticul, cum ar fi terapia de substituție enzimatică (ERT), disponibilă și în România. De asemenea, vom aborda managementul zilnic al bolii și importanța unei abordări multidisciplinare pentru a îmbunătăți calitatea vieții pacienților.
- 🧬 Cauză Genetică: Boala este cauzată de mutații ale genei GAA și se transmite autozomal recesiv, ceea ce înseamnă că un copil trebuie să moștenească o copie a genei defecte de la ambii părinți pentru a dezvolta boala.
- 👶 Forme Clinice: Există o formă infantilă (IOPD), severă, cu afectare cardiacă majoră, și o formă cu debut tardiv (LOPD), care progresează mai lent și afectează în principal mușchii scheletici și respiratori.
- 🔬 Diagnostic: Diagnosticul se bazează pe măsurarea activității enzimei GAA în sânge și este confirmat prin teste genetice.
- 💊 Tratament Revoluționar: Terapia de substituție enzimatică (ERT) cu alglucozidază alfa poate încetini progresia bolii, îmbunătățind semnificativ supraviețuirea și funcția musculară.
Cuprins
🧬 Ce este boala Pompe?
Boala Pompe, denumită și glicogenoză de tip II, este o afecțiune monogenică ereditară, rară și progresivă, care face parte din categoria bolilor de stocaj lizozomal. Aceasta este cauzată de un deficit al enzimei denumite alfa-glucozidaza acidă (GAA). Rolul acestei enzime este de a descompune glicogenul (o formă de zahăr stocată în celule) în glucoză, care este folosită ca sursă de energie. În absența sau la niveluri foarte scăzute ale enzimei GAA, glicogenul se acumulează în interiorul lizozomilor celulelor, ducând la deteriorarea și distrugerea acestora. Afectarea principală este la nivel muscular, vizând mușchii scheletici, respiratori și, în special în formele infantile, mușchiul cardiac.
Frecvența bolii este estimată la aproximativ 1 caz la 40.000 de nașteri, însă aceasta poate varia în funcție de regiunea geografică și etnie. Boala Pompe se manifestă sub forma unui spectru clinic larg, de la forme extrem de severe cu debut în primele luni de viață, până la forme cu debut tardiv, la adulți, cu o progresie mai lentă, dar constantă.
Incidența estimată a bolii
Activitate GAA în forma infantilă
Activitate GAA în forma tardivă
Pacienți LOPD cu debut respirator
🩺 Simptomele bolii Pompe
Manifestările clinice ale bolii Pompe variază semnificativ în funcție de vârsta de debut și de nivelul de activitate enzimatică reziduală. Simptomul comun tuturor formelor este miopatia (slăbiciunea musculară) progresivă.
Simptome comune
Forma Infantilă Clasică (IOPD – Infantile-Onset Pompe Disease)
Debutul are loc în primele luni de viață, fiind cea mai severă formă. Bebelușii par adesea normali la naștere, dar simptomele se dezvoltă rapid:
- Hipotonia musculară severă (floppy baby): Copilul are mușchii foarte moi, dificultăți în a-și susține capul și în a se mișca.
- Cardiomiopatie hipertrofică masivă: Acumularea de glicogen în mușchiul inimii duce la îngroșarea pereților acesteia, afectându-i funcția de pompare. Acesta este simptomul distinctiv al formei infantile.
- Hepatomegalie: Mărirea ficatului.
- Macroglosie: Limba este mărită, ceea ce poate cauza dificultăți de hrănire și respirație.
- Dificultăți de respirație: Slăbiciunea mușchilor respiratori duce la insuficiență respiratorie și infecții pulmonare recurente.
- Eșecul în dezvoltare: Copilul nu ia în greutate și nu atinge etapele motorii normale.
Fără tratament, speranța de viață este, din păcate, mai mică de un an.
Forma cu Debut Tardiv (LOPD – Late-Onset Pompe Disease)
Această formă poate debuta oricând după vârsta de 1 an, în copilărie, adolescență sau chiar la vârsta adultă (între a doua și a șasea decadă de viață). Progresia este mai lentă, iar afectarea cardiacă severă este, de obicei, absentă.
- Slăbiciune musculară progresivă: Afectează predominant mușchii proximali (cei apropiați de trunchi) – șolduri, coapse, umeri și brațe. Pacienții pot avea dificultăți la urcatul scărilor, la ridicarea de pe scaun sau la ridicarea brațelor.
- Afectare respiratorie: Slăbiciunea diafragmului și a altor mușchi respiratori este o caracteristică majoră și adesea prima manifestare. Simptomele includ dispnee (dificultate la respirație) la efort, apnee în somn, infecții respiratorii frecvente și dureri de cap matinale (din cauza hipoventilației nocturne). Aproximativ 13% dintre pacienții cu LOPD prezintă simptome respiratorii la debut.
- Mers legănat (șovăielnic): Din cauza slăbiciunii mușchilor pelvieni.
- Semnul Gowers: Nevoia de a folosi mâinile pentru a “se cățăra” pe propriile picioare pentru a se ridica de la sol.
- Dureri musculare și crampe: În special după efort.
Simptome rare și mai puțin specifice
Pe lângă manifestările clasice, pacienții pot prezenta și alte simptome, care pot face diagnosticul mai dificil:
- Disfagie: Dificultăți la înghițire, din cauza slăbiciunii mușchilor faringieni.
- Pierdere în greutate neintenționată: Cauzată de atrofia musculară și dificultățile de alimentație.
- Tulburări de ritm cardiac (aritmii): Mai frecvente în formele tardive decât se credea inițial.
- Hipoacuzie: Pierderea auzului a fost raportată la unii pacienți.
- Anomalii scheletice: Scolioză sau hiperlordoză lombară (curbură exagerată a spatelui), ca o consecință a slăbiciunii mușchilor paravertebrali.
- Niveluri crescute ale creatinkinazei (CK): Un indicator de leziune musculară, adesea persistent crescut, dar nespecific.
🔬 Cauze și factori de risc
Boala Pompe este o afecțiune pur genetică, cauzată de mutații la nivelul genei GAA, localizată pe cromozomul 17. Această genă conține instrucțiunile pentru producerea enzimei alfa-glucozidaza acidă.
Transmiterea bolii este autozomal recesivă. Aceasta înseamnă că o persoană trebuie să moștenească două copii ale genei GAA modificate (câte una de la fiecare părinte) pentru a dezvolta boala. Părinții care au o singură copie a genei defecte sunt purtători sănătoși; ei nu au simptome, dar pot transmite gena copiilor lor.
Probabilitatea genetică
Dacă ambii părinți sunt purtători ai genei GAA mutante, la fiecare sarcină există:
Factorii de risc sunt exclusiv genetici:
- Istoric familial: Existența unui membru al familiei diagnosticat cu Boala Pompe.
- Consangvinitatea: Căsătoriile între rude de sânge cresc riscul ca ambii părinți să fie purtători ai aceleiași gene recesive.
Nu există factori de mediu, stil de viață sau dietă care să cauzeze boala.
Tipuri de Boala Pompe
Spectrul bolii Pompe este continuu, dar pentru scopuri clinice, este împărțit în două categorii principale, bazate pe vârsta de debut și severitatea afectării cardiace.
| Tipul Bolii | Vârsta de Debut | Severitate | Caracteristici Cheie | Activitate Enzimatică GAA |
|---|---|---|---|---|
| Forma Infantilă Clasică (IOPD) | < 12 luni | Severă, progresie rapidă | Cardiomiopatie hipertrofică severă, hipotonie marcată (“floppy baby”), insuficiență cardio-respiratorie. Fără tratament, decesul survine în primul an de viață. | < 1% din nivelul normal |
| Forma cu Debut Tardiv (LOPD) | > 1 an (copilărie, adolescență, adult) | Ușoară spre severă, progresie lentă | Slăbiciune musculară proximală progresivă, afectare respiratorie predominantă, afectarea cardiacă este de obicei absentă sau minoră. Evoluția duce la dizabilitate și dependență de scaun rulant/ventilație. | 1% – 30% din nivelul normal |
Diagnosticarea bolii Pompe
Diagnosticul precoce al bolii Pompe este crucial pentru a iniția tratamentul cât mai devreme și a încetini progresia bolii. Procesul de diagnosticare implică mai mulți pași.
-
Suspiciune clinicăSe bazează pe prezența simptomelor caracteristice: slăbiciune musculară, probleme respiratorii inexplicabile, cardiomiopatie la sugari sau niveluri persistent crescute de CK.
-
Testarea activității enzimaticeAceasta este principala metodă de diagnostic. Se măsoară activitatea enzimei GAA dintr-o probă de sânge uscat (DBS – Dried Blood Spot) sau din sânge venos. Un nivel scăzut sau absent al activității GAA confirmă suspiciunea.
-
Confirmare geneticăAnaliza moleculară a genei GAA este esențială pentru a confirma diagnosticul. Aceasta identifică mutațiile specifice care cauzează boala și poate oferi informații despre prognostic.
Alte investigații care pot sprijini diagnosticul și evalua gradul de afectare includ:
- Electromiografia (EMG): Poate arăta semne de miopatie, adesea cu descărcări miotonice, indicând o “iritabilitate” a membranei musculare.
- Biopsia musculară: În trecut era o metodă standard, dar acum este mai rar utilizată. Aceasta poate evidenția acumularea de glicogen în vacuolele din fibrele musculare.
- Teste imagistice: RMN muscular pentru a evalua distribuția afectării musculare, ecocardiografie pentru a detecta cardiomiopatia și teste funcționale pulmonare pentru a măsura capacitatea respiratorie.
În unele țări, screeningul neonatal pentru Boala Pompe este implementat, permițând diagnosticarea și tratarea bolii înainte de apariția simptomelor severe. În România, există proiecte pilot în acest sens.
Când să consulți un medic
Semnale de alarmă
Adresează-te unui medic dacă tu sau copilul tău prezentați oricare dintre următoarele simptome, mai ales dacă acestea sunt progresive:
- La sugari: hipotonie musculară evidentă (copil “moale”), dificultăți de hrănire, nu ia în greutate, respirație dificilă sau rapidă.
- La copii și adulți: slăbiciune musculară inexplicabilă care se agravează în timp (dificultăți la urcatul scărilor, ridicarea de obiecte), dificultăți de respirație la efort sau în poziție culcat, dureri de cap dimineața, căderi frecvente sau infecții pulmonare recurente.
Tratament pentru Boala Pompe
Până de curând, managementul bolii Pompe era exclusiv de susținere. Apariția Terapiei de Substituție Enzimatică (ERT) a revoluționat prognosticul, în special pentru forma infantilă.
Medicamentul utilizat este alglucozidaza alfa (disponibil sub denumirile comerciale Myozyme® pentru pacienții pediatrici și Lumizyme® pentru cei adulți). Cochrane a concluzionat că ERT poate încetini progresia bolii și îmbunătăți calitatea vieții pacienților cu forma tardivă (LOPD), comparativ cu placebo. În România, acest tratament este disponibil gratuit pentru pacienții cu diagnostic confirmat, prin Programul Național de Tratament pentru Boli Rare.
Prognostic IOPD FĂRĂ ERT
• Progresie rapidă
• Insuficiență cardiacă severă
• Deces în primul an de viață
Prognostic IOPD CU ERT
• Supraviețuire extinsă (>5 ani)
• Îmbunătățirea funcției cardiace
• Achiziții motorii posibile
Avantajele ERT
- Îmbunătățește supraviețuirea în forma infantilă.
- Stabilizează sau încetinește declinul funcției musculare și respiratorii în LOPD.
- Reduce dimensiunea inimii (cardiomegalia) la sugari.
- Poate îmbunătăți calitatea vieții și toleranța la efort.
Considerații și provocări
- Tratament pe viață, administrat prin perfuzii la fiecare 2 săptămâni.
- Eficacitatea poate varia în funcție de severitatea bolii la debut și de statusul imunologic al pacientului.
- Posibile reacții la perfuzie sau dezvoltarea de anticorpi împotriva enzimei.
- Nu vindecă boala, ci o gestionează.
Terapii de susținere
O abordare multidisciplinară este esențială:
- Suport respirator: Ventilație non-invazivă (BiPAP) în timpul nopții pentru a combate hipoventilația și apneea în somn. În stadii avansate, poate fi necesară traheostomia și ventilația invazivă.
- Fizioterapie și kinetoterapie: Pentru a menține forța musculară, flexibilitatea articulațiilor și pentru a preveni contracturile.
- Terapie ocupațională: Pentru adaptarea activităților zilnice și utilizarea de dispozitive ajutătoare.
- Management nutrițional: O dietă echilibrată, bogată în proteine și carbohidrați complecși, adaptată pentru a preveni pierderea în greutate și a susține masa musculară. Poate fi necesară hrănirea prin gastrostomă în caz de disfagie severă.
- Logopedie: Pentru managementul dificultăților de vorbire și înghițire.
Terapii emergente
Cercetarea continuă este intensă. Terapiile genice, care vizează corectarea defectului genetic de bază, se află în studii clinice avansate și reprezintă o mare speranță pentru viitor.
Managementul bolii și stil de viață
Managementul pe termen lung al bolii Pompe necesită o monitorizare regulată și o colaborare strânsă între pacient și o echipă medicală formată din neurolog, cardiolog, pneumolog, gastroenterolog, dietetician și fizioterapeut.
- Monitorizare regulată: Pacienții trebuie să efectueze evaluări cardiace (EKG, ecocardiografie) și respiratorii (teste funcționale pulmonare, studii de somn) periodice pentru a detecta și trata complicațiile devreme.
- Activitate fizică: Exercițiile fizice de intensitate mică spre moderată, cum ar fi înotul sau mersul pe jos, sunt încurajate pentru a menține tonusul muscular, dar efortul extrem trebuie evitat.
- Dietă: O alimentație bogată în proteine poate ajuta la susținerea masei musculare. Carbohidrații complecși oferă o sursă de energie constantă. Suplimentele pot fi necesare, la recomandarea medicului.
Diferențele de management se bazează mai mult pe tipul bolii decât pe sex:
- IOPD: Prioritatea absolută este inițierea ERT cât mai precoce posibil, ideal în primele săptămâni de viață, și managementul intensiv al funcției cardiace.
- LOPD: Accentul cade pe monitorizarea funcției respiratorii și pe inițierea suportului ventilator la momentul optim, alături de fizioterapie constantă.
🧠 Testează-ți cunoștințele
Care este principalul tratament specific care a schimbat prognosticul bolii Pompe?
Complicații posibile
Fiind o boală progresivă, Boala Pompe poate duce la o serie de complicații grave, care afectează calitatea și durata vieții:
- Insuficiență respiratorie: Este principala cauză de morbiditate și mortalitate în forma cu debut tardiv. Slăbiciunea mușchilor respiratori duce la incapacitatea de a respira eficient, necesitând suport ventilator permanent.
- Insuficiență cardiacă: Este complicația dominantă și fatală în forma infantilă netratată.
- Dizabilitate motorie severă: Progresia slăbiciunii musculare duce la pierderea capacității de a merge, necesitând utilizarea unui scaun cu rotile.
- Complicații ortopedice: Contracturi articulare, scolioză severă, osteoporoză din cauza imobilității.
- Malnutriție și probleme de creștere: Din cauza dificultăților de hrănire și a consumului energetic ridicat.
Prevenție
Deoarece boala Pompe este o afecțiune genetică, prevenția primară nu este posibilă. Însă, prevenția transmiterii la generațiile viitoare se poate realiza prin:
- Sfat genetic (consiliere genetică): Familiile cu istoric de boală Pompe pot beneficia de consiliere pentru a înțelege riscurile și opțiunile disponibile.
- Screeningul purtătorilor: Membrii familiei unui pacient pot fi testați pentru a vedea dacă sunt purtători ai genei mutante.
- Diagnosticul prenatal: Pentru cuplurile cu risc (ambii părinți purtători), se poate testa fătul în timpul sarcinii prin amniocenteză sau biopsia vilozităților coriale pentru a determina dacă este afectat.
Întrebări frecvente
▼
Este considerată o boală rară, cu o incidență estimată de 1 la 40.000 de nou-născuți. Totuși, se crede că forma cu debut tardiv este subdiagnosticată, iar numărul real de cazuri ar putea fi mai mare.
▼
În prezent, nu există un leac pentru Boala Pompe. Cu toate acestea, terapia de substituție enzimatică (ERT) poate încetini semnificativ progresia bolii, poate ameliora simptomele și poate prelungi viața, transformând-o dintr-o boală rapid fatală (în cazul formei infantile) într-o afecțiune cronică, gestionabilă.
▼
Prognosticul este variabil și depinde de rata de progresie a bolii. Este o boală progresivă, care duce în timp la dizabilitate semnificativă. Afectarea respiratorie este principalul factor care influențează speranța de viață. Studiile arată că aproximativ 30% dintre pacienții cu LOPD devin dependenți de ventilație mecanică de-a lungul vieții. Tratamentul cu ERT poate stabiliza funcția respiratorie și musculară pentru perioade lungi de timp.
Referințe / Surse
Cleveland Clinic. (n.d.). Pompe Disease: Symptoms & Treatment. my.clevelandclinic.org/health/diseases/15808-pompe-disease
Cupler, E. J., et al. (2012). S1.3 Adult-onset Pompe disease. PMC. pmc.ncbi.nlm.nih.gov/articles/PMC3298100/
Lurie Children’s Hospital of Chicago. (n.d.). Pompe Disease. www.luriechildrens.org/en/specialties-conditions/pompe-disease/
National Organization for Rare Disorders (NORD). (n.d.). Pompe Disease. rarediseases.org/rare-diseases/pompe-disease/
van der Ploeg, A. T., & Reuser, A. J. (2008). Clinical features of Pompe disease. PMC. pmc.ncbi.nlm.nih.gov/articles/PMC3866902/




