


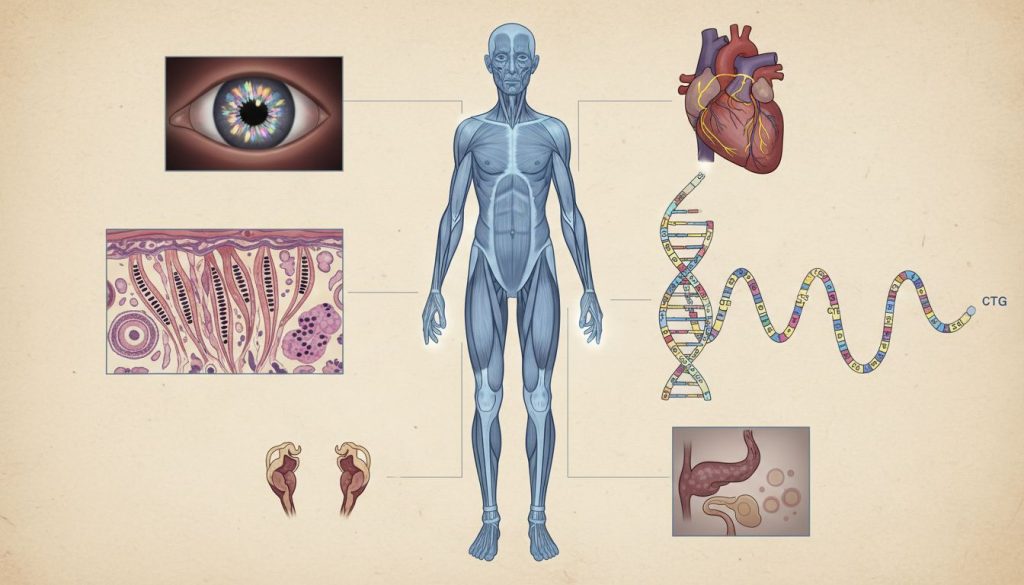
Distrofia miotonică de tip 1 (DM1), cunoscută și ca boala Steinert, este cea mai frecventă formă de distrofie musculară care debutează la vârsta adultă. Această afecțiune genetică complexă se manifestă printr-o combinație unică de miotonie (dificultatea de a relaxa mușchii după contracție) și slăbiciune musculară progresivă, care afectează nu doar musculatura, ci și inima, ochii, sistemul endocrin și cogniția. Boala este cauzată de o mutație în gena DMPK de pe cromozomul 19 și se transmite autosomal dominant, ceea ce înseamnă că un copil are un risc de 50% să moștenească boala de la un părinte afectat.
Articolul explorează în detaliu toate aspectele bolii Steinert, de la tipurile clinice (clasic și congenital) și simptomele specifice, la metodele de diagnostic, opțiunile de management al bolii și complicațiile posibile. Deși nu există un tratament care să vindece boala, managementul simptomatic prin fizioterapie, medicamente, monitorizare cardiacă și suport respirator poate îmbunătăți semnificativ calitatea vieții și poate încetini progresia unor manifestări.
- 🧬 Cauză Genetică: O expansiune a repetițiilor trinucleotidice (CTG) în gena DMPK. Severitatea bolii se corelează cu numărul de repetiții.
- 🖐️ Simptome Cheie: Miotonie (pumn care nu se poate deschide rapid), slăbiciune la nivelul feței, gâtului și membrelor, ducând la “fața miotonică” și dificultăți la mers.
- ❤️ Impact Sistemic: Afectează multiple organe, cu risc de cataractă, aritmii cardiace, tulburări digestive și probleme endocrine.
- 👶 Forme Clinice: Există o formă clasică cu debut la adult (20-30 de ani) și una congenitală, mult mai severă, prezentă de la naștere.
- 💊 Management: Nu există leac, dar tratamentul se concentrează pe gestionarea simptomelor: medicamente pentru miotonie, pacemaker pentru inimă, fizioterapie și consiliere genetică.
Cuprins
🧬 Despre Distrofia miotonică Steinert
Distrofia miotonică de tip 1 (DM1), cunoscută și sub denumirea de boala Steinert, este o afecțiune neuromusculară complexă și ereditară. Este considerată cea mai frecventă formă de distrofie musculară diagnosticată la adulți, afectând aproximativ 1 din 8.000 de persoane la nivel global, fără predilecție pentru o anumită etnie. Boala este definită de două caracteristici principale: miotonie, care reprezintă incapacitatea mușchilor de a se relaxa rapid după o contracție, și o slăbiciune musculară progresivă (atrofie), care avansează lent de-a lungul anilor.
Spre deosebire de alte distrofii musculare care afectează predominant mușchii scheletici, boala Steinert are un impact multisistemic. Aceasta înseamnă că poate afecta o gamă largă de organe și sisteme, incluzând inima, ochii, sistemul digestiv, sistemul endocrin și sistemul nervos central. Din cauza acestei naturi complexe, managementul pacienților necesită o abordare multidisciplinară. Boala este transmisă genetic, printr-un model autosomal dominant, ceea ce înseamnă că o singură copie a genei modificate (moștenită de la unul dintre părinți) este suficientă pentru a cauza boala. Debutul simptomelor are loc, de obicei, la vârsta adultă tânără, între 20 și 30 de ani, deși există și forme severe, congenitale.
Prevalență globală
Risc de transmitere la copil
Vârsta tipică a debutului
Sisteme de organe afectate
🩺 Ce este? Tipuri de Distrofie miotonică
Distrofia miotonică Steinert variază considerabil în severitate și vârstă de debut, chiar și în cadrul aceleiași familii. Această variabilitate a dus la clasificarea bolii în mai multe forme clinice, cele mai importante fiind forma clasică și forma congenitală.
Forma clasică (adultă)
Aceasta este cea mai comună prezentare a bolii Steinert, cu simptome care apar de obicei la adolescență sau la vârsta adultă tânără (între 20 și 30 de ani). Progresia este, în general, lentă. Pacienții dezvoltă triada clasică de simptome:
- Miotonie: Dificultate în relaxarea mușchilor, resimțită cel mai adesea la nivelul mâinilor. De exemplu, după ce strâng pumnul sau apucă o clanță, pacienții nu pot deschide mâna imediat.
- Atrofie și slăbiciune musculară progresivă: Afectează inițial mușchii feței (ptoza palpebrală – pleoape căzute, obraji supți), mușchii gâtului și mușchii distali ai membrelor (antebrațe, gambe).
- Afectare multisistemică: Pe parcursul anilor, apar și alte probleme, cum ar fi cataracta (adesea la o vârstă neobișnuit de tânără), tulburări de ritm cardiac și probleme endocrine.
Forma congenitală (severă)
Aceasta este cea mai gravă formă de DM1 și este prezentă de la naștere. Spre deosebire de forma clasică, aceasta este aproape întotdeauna moștenită de la mamă. Nou-născuții prezintă hipotonie severă (sunt “moi”, ca o păpușă de cârpă), dificultăți respiratorii majore și probleme de hrănire. Miotonia clinică este de obicei absentă la această vârstă, dar slăbiciunea musculară este profundă. Rata mortalității în primul an de viață este ridicată (aproximativ 20-30%), principala cauză fiind insuficiența respiratorie. Copiii care supraviețuiesc se confruntă cu întârzieri semnificative în dezvoltarea motorie și dizabilități intelectuale.
Forma Clasică (Adultă)
• Debut: 20-30 ani
• Simptom principal inițial: Miotonie
• Progresie: Lentă, pe zeci de ani
• Afectare cognitivă: Ușoară spre moderată
Forma Congenitală
• Debut: De la naștere
• Simptom principal inițial: Hipotonie severă
• Progresie: Risc vital în perioada neonatală
• Afectare cognitivă: Moderată spre severă
🌡️ Simptome comune
Manifestările bolii Steinert sunt diverse și afectează progresiv calitatea vieții. Ele pot fi împărțite în simptome musculare și sistemice.
Simptome musculare
- Miotonie de acțiune și percuție: Fenomenul definitoriu al bolii. Se manifestă ca o rigiditate tranzitorie a mușchilor după o contracție voluntară. Este frecventă la nivelul mâinilor și antebrațelor (dificultate la eliberarea obiectelor, la scris), dar poate afecta și mușchii feței sau ai limbii, ducând la o vorbire disartrică, nazonată. Miotonia este adesea agravată de frig și poate fi ameliorată prin mișcări repetitive (“warm-up phenomenon”).
-
Slăbiciune musculară progresivă și atrofie: Slăbiciunea musculară nu este uniformă. Afectează în principal:
- Mușchii feței și gâtului: Duce la un aspect facial distinctiv, numit “mască miotonică” sau “facies miotonic”, caracterizat prin ptoză palpebrală (pleoape căzute), obraji supți și o expresie tristă sau indiferentă. Slăbiciunea mușchilor gâtului face dificilă ridicarea capului de pe pernă.
- Mușchii distali ai membrelor: Slăbiciunea antebrațelor și a mâinilor afectează prehensiunea, iar slăbiciunea mușchilor gambei duce la picior căzut (steppage gait), provocând un mers șovăielnic și un risc crescut de cădere.
- Mușchii respiratori: Slăbiciunea diafragmului și a mușchilor intercostali poate duce la insuficiență respiratorie, în special în timpul somnului.
Simptome sistemice
Boala Steinert este o afecțiune multisistemică, ceea ce înseamnă că efectele sale se extind dincolo de mușchi.
-
Afectare ocularăCataracta este extrem de frecventă, apărând adesea la vârste tinere (30-40 de ani) și având un aspect specific (opacități policrome, “în pom de Crăciun”). Ptoza palpebrală este de asemenea comună.
-
Afectare cardiacăEste una dintre cele mai serioase complicații. Peste 80% dintre pacienți dezvoltă anomalii pe electrocardiogramă (ECG), cel mai adesea tulburări de conducere (blocuri atrioventriculare). Acestea pot duce la bradicardie, sincope (leșin) și, în cazuri grave, la moarte subită. De aceea, monitorizarea cardiologică regulată este esențială.
-
Afectare respiratorieSlăbiciunea mușchilor respiratori duce la hipoventilație, în special noaptea, provocând apnee în somn și acumularea de dioxid de carbon în sânge. Aceasta este o cauză majoră de morbiditate și mortalitate.
-
Afectare endocrinăSunt frecvente disfuncțiile hormonale. Bărbații pot suferi de atrofie testiculară, hipogonadism și infertilitate. Femeile pot avea cicluri neregulate. Rezistența la insulină este de asemenea comună.
-
Afectare cognitivăMulte persoane cu DM1 se confruntă cu oboseală cronică extremă și somnolență diurnă excesivă. De asemenea, pot apărea deficite cognitive, incluzând dificultăți de planificare, apatie și probleme de personalitate.
🧐 Simptome rare
Pe lângă manifestările comune, există și o serie de simptome mai puțin frecvente, dar care pot avea un impact semnificativ asupra pacienților:
- Alopecie frontală prematură: În special la bărbați, căderea părului în zona frontală poate fi un semn timpuriu al bolii.
- Probleme gastrointestinale: Unii pacienți pot suferi de constipație cronică, diaree, sau disfagie (dificultăți la înghițire) din cauza afectării mușchilor netezi ai tractului digestiv.
- Scolioză: Deformarea coloanei vertebrale poate apărea, în special la cei cu debut timpuriu al bolii.
- Dureri musculare: Deși nu este un simptom universal, unii pacienți raportează dureri cronice la nivelul mușchilor.
🔬 Cauze și factori de risc
Distrofia miotonică Steinert este o boală pur genetică. Nu este cauzată de stilul de viață, dietă sau factori de mediu.
Cauza principală este o mutație de tip expansiune în gena DMPK (Dystrophia Myotonica Protein Kinase), localizată pe cromozomul 19 (mai exact, în regiunea 19q13.3). În mod normal, această genă conține o secvență de trei nucleotide (Citozină-Timină-Guanină sau CTG) care se repetă de 5 până la 37 de ori. La persoanele cu DM1, această secvență este instabilă și se repetă de un număr anormal de mare de ori (de la 50 până la câteva mii).
Moștenire și Risc
- Transmitere autosomal dominantă: O persoană afectată are un risc de 50% de a transmite gena defectă la fiecare sarcină, indiferent de sexul copilului.
- Penetranță 100%: Orice persoană care moștenește expansiunea va dezvolta boala, dar severitatea și vârsta de debut variază (expresivitate variabilă).
Fenomenul de Anticipare
- Numărul de repetiții CTG tinde să crească de la o generație la alta, în special când este transmis de la mamă.
- Acest lucru duce la fenomenul de anticipare genetică: boala apare la o vârstă mai timpurie și cu simptome mai severe în generațiile succesive.
❤️ Diagnosticarea
Diagnosticul bolii Steinert se bazează pe o combinație de evaluare clinică, teste electrofiziologice și confirmare genetică. Procesul de diagnosticare este esențial pentru a exclude alte afecțiuni neuromusculare și pentru a iniția un plan de management adecvat.
-
1. Anamneză și examen clinic: Medicul neurolog va evalua istoricul familial și va căuta semnele clinice caracteristice: miotonie (poate fi demonstrată cerând pacientului să strângă pumnul), slăbiciune și atrofie musculară cu distribuție specifică (față, gât, membre distale), și semne sistemice precum cataracta.
-
2. Electromiografie (EMG): Acesta este un test cheie care înregistrează activitatea electrică a mușchilor. La pacienții cu DM1, EMG-ul relevă un model specific numit “descărcare miotonică” – un sunet caracteristic, asemănător cu cel al unui bombardier în picaj, care confirmă prezența miotoniei la nivel electric, chiar dacă nu este evidentă clinic.
-
3. Testare genetică: Acesta este standardul de aur pentru confirmarea diagnosticului. O probă de sânge este analizată pentru a măsura numărul de repetiții CTG în gena DMPK. Un număr de peste 50 de repetiții confirmă diagnosticul de DM1.
-
4. Evaluări suplimentare: Odată confirmat diagnosticul, sunt necesare investigații pentru a evalua afectarea sistemică:
- Electrocardiogramă (ECG) și Holter ECG 24h: Pentru a detecta aritmiile și tulburările de conducere cardiacă.
- Examen oftalmologic: Pentru a diagnostica prezența cataractei.
- Teste de sânge: Pentru a verifica nivelurile hormonale și glicemia.
- Teste funcționale respiratorii: Pentru a evalua funcția pulmonară.
-
5. Biopsia musculară (rar utilizată): În trecut, era o metodă frecventă. Astăzi, se realizează rar, doar în cazuri atipice, deoarece testarea genetică este mult mai precisă.
👨⚕️ Când să consulți un medic
Semne de alarmă
Este crucial să consulți un medic neurolog dacă prezinți unul sau mai multe dintre următoarele simptome, mai ales dacă există un istoric familial de boli musculare:
- Dificultate persistentă în a relaxa mâna după ce ai strâns un obiect.
- Slăbiciune la nivelul feței (pleoape căzute), gâtului sau picioarelor (mers împiedicat).
- Palpitații, amețeli inexplicabile sau episoade de leșin (sincope).
- Diagnostic de cataractă la o vârstă neobișnuit de tânără (sub 50 de ani).
- Oboseală extremă și somnolență în timpul zilei care nu se ameliorează cu odihnă.
💊 Tratament și management
În prezent, nu există un tratament care să vindece distrofia miotonică Steinert sau să oprească progresia acesteia. Cu toate acestea, există numeroase strategii de management care pot ameliora simptomele, pot preveni complicațiile și pot îmbunătăți semnificativ calitatea vieții.
Abordare multidisciplinară
- Managementul miotoniei: Pentru pacienții la care miotonia este severă și dizabilitantă, se pot prescrie medicamente antiaritmice precum Mexiletina. Acestea ajută la stabilizarea membranei celulare musculare, reducând rigiditatea.
- Managementul cardiac: Din cauza riscului de tulburări de conducere, monitorizarea cardiologică anuală cu ECG este obligatorie. În cazul în care se detectează blocuri cardiace semnificative, poate fi necesară implantarea unui pacemaker sau a unui defibrilator cardioverter implantabil (ICD) pentru a preveni moartea subită.
- Managementul respirator: Pacienții trebuie evaluați periodic pentru probleme respiratorii. În cazul hipoventilației nocturne sau a apneei în somn, se recomandă utilizarea unui aparat de ventilație non-invazivă (BiPAP) pe timpul nopții.
- Fizioterapie și Terapie ocupațională: Exercițiile fizice moderate, adaptate capacității pacientului, sunt esențiale pentru a menține forța musculară, flexibilitatea articulațiilor și pentru a preveni contracturile. Terapia ocupațională ajută pacienții să se adapteze la limitările fizice și să-și mențină independența în activitățile zilnice.
- Managementul oftalmologic: Cataracta se tratează chirurgical, prin înlocuirea cristalinului opacifiat.
- Terapii emergente: Cercetarea este foarte activă. Terapiile viitoare, precum cele bazate pe oligonucleotide antisens (ASO) sau editarea genetică (CRISPR), care țintesc direct mecanismul molecular al bolii, sunt în diverse stadii de studiu clinic și reprezintă o speranță majoră pentru viitor.
🧘 Stil de viață și remedii la domiciliu
Adaptarea stilului de viață poate juca un rol important în gestionarea simptomelor și menținerea unei calități bune a vieții.
✅ Ce ajută (Do’s)
- Exerciții fizice moderate: Mersul pe jos, înotul sau ciclismul staționar pot ajuta la menținerea tonusului muscular fără a epuiza organismul. Evitați efortul extenuant.
- Menținerea unei greutăți sănătoase: Greutatea în exces pune o presiune suplimentară pe mușchii slăbiți și pe articulații.
- Dietă bogată în fibre: Pentru a combate constipația, se recomandă o dietă bogată în fructe, legume și cereale integrale, alături de o hidratare adecvată.
- Planificarea activităților: Gestionarea energiei este crucială. Planificați sarcinile dificile pentru momentele zilei în care vă simțiți cel mai odihnit.
❌ Ce trebuie evitat (Don’ts)
- Expunerea la frig: Temperaturile scăzute agravează semnificativ miotonia. Îmbrăcați-vă adecvat pe vreme rece.
- Anumite medicamente: Unele medicamente, în special anumite anestezice și relaxante musculare, pot fi periculoase pentru persoanele cu DM1. Este esențial ca orice medic să fie informat despre diagnostic înainte de orice procedură.
- Alcoolul: Consumul de alcool poate agrava somnolența și slăbiciunea musculară.
- Ignorarea oboselii: Forțarea limitelor poate duce la epuizare și la un risc crescut de accidente.
🚻 Diferențe de tratament în funcție de sex/vârstă
Managementul bolii Steinert necesită o atenție specială la particularitățile legate de vârstă și sex.
- Bărbați: Atrofia testiculară și hipogonadismul pot necesita terapie de substituție cu testosteron pentru a îmbunătăți libidoul, energia și densitatea osoasă. Fertilitatea poate fi compromisă, deci consilierea pe această temă este importantă.
- Femei: Sarcina la femeile cu DM1 este considerată una cu risc ridicat. Există un risc crescut de complicații obstetricale (avort spontan, naștere prematură) și un risc de a naște un copil cu forma congenitală severă. Este necesară o monitorizare atentă de către o echipă multidisciplinară.
- Copii (forma congenitală): Managementul este intensiv de la naștere. Suportul ventilatoriu, hrănirea prin sondă nazogastrică și terapiile de stimulare a dezvoltării (fizioterapie, logopedie, terapie ocupațională) sunt esențiale pentru supraviețuire și pentru a maximiza potențialul copilului.
⚠️ Complicații posibile
Progresia lentă a bolii poate duce la o serie de complicații grave, care reprezintă principalele cauze de morbiditate și mortalitate.
Complicații majore
🛡️ Prevenție
Deoarece distrofia miotonică Steinert este o boală genetică, prevenția primară (adică împiedicarea apariției bolii) nu este posibilă. Prevenția se concentrează pe opțiunile reproductive pentru persoanele afectate și pe prevenirea complicațiilor bolii.
- Consiliere genetică: Este crucială pentru persoanele cu DM1 și familiile acestora. Un consilier genetic poate explica modelul de moștenire, riscul de recurență în familie și opțiunile reproductive disponibile.
- Testare prenatală: Pentru cuplurile în care unul dintre parteneri are DM1, există opțiuni pentru a testa fătul în timpul sarcinii (prin amniocenteză sau biopsia vilozităților coriale).
- Diagnostic genetic preimplantator (PGD): Aceasta este o opțiune pentru cuplurile care optează pentru fertilizarea in vitro (FIV). Embrionii sunt testați genetic înainte de a fi implantați în uter, permițând selectarea celor care nu au moștenit mutația.
🧠 Verifică-ți cunoștințele
Care este fenomenul genetic responsabil pentru apariția simptomelor mai devreme și mai severe în generațiile succesive ale persoanelor cu distrofie miotonică Steinert?
❓ Întrebări frecvente
▼
Progresia este foarte variabilă. În forma clasică, slăbiciunea musculară avansează lent, pe parcursul a zeci de ani. Speranța de viață este redusă, cu o medie între 50 și 60 de ani, dar depinde enorm de severitatea afectării cardiace și respiratorii. Cu o monitorizare atentă și un management proactiv al complicațiilor, mulți pacienți pot trăi mai mult.
▼
Nu, în prezent nu există un tratament care să vindece boala. Tratamentele actuale sunt simptomatice și vizează gestionarea manifestărilor bolii (miotonia, problemele cardiace, respiratorii etc.) pentru a îmbunătăți calitatea și durata vieții. Cercetările active în domeniul terapiilor genetice oferă speranțe pentru viitor.
▼
Da, exercițiile fizice de intensitate ușoară până la moderată sunt chiar recomandate. Activități precum înotul, mersul pe jos sau ciclismul pot ajuta la menținerea forței musculare, a funcției cardiovasculare și a stării generale de bine. Este important să se evite efortul extenuant, care poate duce la oboseală excesivă și la deteriorarea mușchilor. Este ideal să discutați cu un fizioterapeut pentru a stabili un program de exerciții personalizat.
📚 Resurse și informații suplimentare
Pentru informații detaliate și resurse actualizate, puteți consulta site-urile organizațiilor internaționale dedicate bolilor neuromusculare și distrofiei miotonice. Acestea oferă ghiduri pentru pacienți, informații despre studii clinice și suport pentru familii.
Myotonic Dystrophy Foundation. (n.d.). DM1. myotonic.org/what-is-dm/dm1
National Institute of Neurological Disorders and Stroke (NINDS). (2023). Myotonic Dystrophy Fact Sheet. ninds.nih.gov/health-information/disorders/myotonic-dystrophy
Muscular Dystrophy Association (MDA). (n.d.). Myotonic Dystrophy Type 1. mda.org/disease/myotonic-dystrophy/types/myotonic-dystrophy-type-1
Johns Hopkins Medicine. (n.d.). Myotonic Dystrophy. hopkinsmedicine.org/health/conditions-and-diseases/myotonic-dystrophy
Mayo Clinic. (2022). Myotonic dystrophy. mayoclinic.org/diseases-conditions/myotonic-dystrophy/symptoms-causes/syc-20354601




