


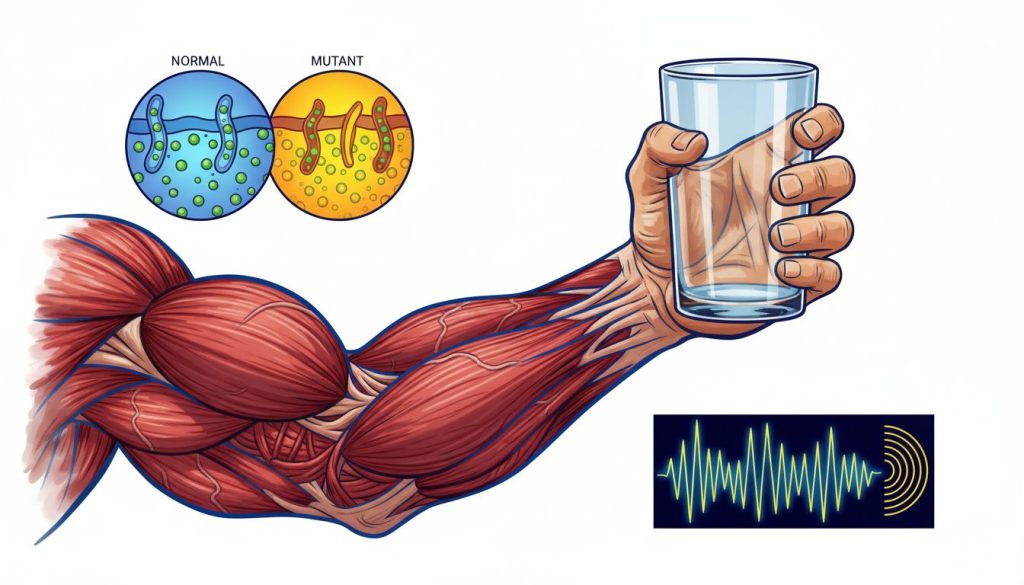
Miotonia congenitală Thomsen este o afecțiune neuromusculară rară, moștenită genetic, ce se manifestă printr-o relaxare musculară întârziată după o contracție voluntară. Deși nu poate fi vindecată, simptomele sale, precum rigiditatea musculară exacerbată de frig, pot fi gestionate eficient prin tratament medicamentos și adaptarea stilului de viață, permițând pacienților să ducă o viață normală. Diagnosticul se bazează pe simptomele clinice, electromiografie (EMG) și este confirmat prin testare genetică a genei CLCN1.
- 🧬 Cauză Genetică: Afecțiunea este cauzată de mutații în gena CLCN1, transmisă autosomal dominant. Asta înseamnă că o singură copie a genei mutante de la un părinte este suficientă pentru a dezvolta boala.
- 💪 Simptom Principal: Relaxarea lentă și dificilă a mușchilor după efort (ex: imposibilitatea de a descleșta rapid pumnul), cunoscută sub numele de miotonie. Nu este asociată cu slăbiciune musculară progresivă.
- ❄️ Factori Agravanți: Simptomele sunt adesea declanșate sau înrăutățite de frig și de perioadele de inactivitate.
- 💊 Management: Tratamentul include medicamente (precum mexiletina) pentru a reduce rigiditatea și strategii de “încălzire” a mușchilor înainte de activitate fizică.
- 👍 Prognostic Pozitiv: Boala Thomsen este considerată o formă benignă de miotonie, neafectând speranța de viață și având un impact redus asupra calității vieții atunci când este gestionată corespunzător.
Cuprins
ℹ️ Ce este miotonia congenitală Thomsen?
Miotonia congenitală Thomsen, denumită și boala Thomsen, este o afecțiune neuromusculară ereditară, non-distrofică, ceea ce înseamnă că nu provoacă o degradare progresivă (atrofie) a mușchilor, spre deosebire de alte boli precum distrofia musculară Steinert [3]. Caracteristica fundamentală a acestei boli este miotonia, o anomalie musculară care se manifestă printr-o decontractare anormal de lentă a mușchilor după o contracție voluntară [6, 7].
Pacienții descriu acest fenomen ca o rigiditate sau o “înțepenire” temporară. De exemplu, după ce strâng pumnul cu putere, le este foarte greu să-și desfacă degetele imediat [7]. Această rigiditate tinde să se amelioreze după câteva repetări ale mișcării, un fenomen cunoscut sub numele de “warm-up” (încălzire).
Boala este cauzată de mutații în gena CLCN1, localizată pe cromozomul 7, care codifică un canal de clorură esențial pentru funcționarea normală a fibrelor musculare scheletice [10]. Aceste canale ajută la “resetarea” electrică a mușchiului după o contracție. Când funcționează defectuos, mușchii rămân hiperexcitabili și se contractă prelungit. Miotonia congenitală Thomsen are un model de transmitere autosomal dominant [1].
Prevalența Globală
Cazuri cauzate de gena CLCN1
Simptome predominante în picioare/mâini
% îmbunătățire cu Mexiletină
Deși are o prevalență globală estimată la 1 la 100.000 de persoane, este semnificativ mai frecventă în țările scandinave, precum Norvegia, Suedia și Finlanda, datorită unor efecte genetice fondatoare [1].
🌡️ Simptomele bolii Thomsen
Simptomatologia în miotonia congenitală Thomsen debutează de obicei în copilăria timpurie sau chiar la naștere și variază ca intensitate de la un pacient la altul [1, 2]. Principalul simptom este rigiditatea musculară nedureroasă (spasm tonic), care apare după o perioadă de repaus și se ameliorează cu mișcarea.
Simptome comune
- Rigiditate musculară (miotonia): Dificultate în relaxarea mușchilor după ce au fost contractați. Acest lucru se manifestă cel mai frecvent la nivelul mâinilor, picioarelor și pleoapelor. Activități simple devin provocatoare [1, 2]:
- Dificultatea de a elibera un obiect strâns în mână.
- Dificultatea de a deschide ochii rapid după un strănut sau după plâns.
- Probleme la ridicarea de pe un scaun sau din pat după o perioadă de inactivitate.
- Rigiditate la primii pași după ce a stat jos.
- Dificultăți la urcatul scărilor.
- Fenomenul de “încălzire” (Warm-up): Rigiditatea scade și mișcările devin mai fluide după câteva repetări. De exemplu, primii pași pot fi rigizi și lenți, dar după câțiva metri, mersul devine aproape normal.
- Hipertrofie musculară: Pacienții dezvoltă adesea o masă musculară proeminentă, în special la nivelul coapselor, gambelor și umerilor, chiar și în absența unui antrenament fizic intens. Acest aspect atletic este uneori primul semn care atrage atenția [1, 2].
- Agravarea la frig: Temperaturile scăzute exacerbează semnificativ miotonia. Pentru mulți pacienți, expunerea la frig poate face mișcările aproape imposibile pentru scurt timp [1].
- Oboseală post-efort: Deși mușchii sunt puternici, efortul prelungit poate duce la o stare de oboseală musculară.
Simptome rare și specifice
- Spasme faciale și la nivelul trunchiului: Deși mai puțin frecvente, miotonia poate afecta și mușchii feței (de exemplu, mușchii implicați în masticație) sau ai trunchiului [1].
- Afectare predominantă a membrelor inferioare: La unii pacienți, simptomele sunt mult mai pronunțate la nivelul picioarelor decât la mâini.
- Slăbiciune tranzitorie: Spre deosebire de forma Becker, slăbiciunea musculară nu este o caracteristică definitorie a bolii Thomsen. Totuși, unii pacienți pot experimenta episoade scurte și ocazionale de slăbiciune, în special după un efort brusc și intens [2].
🧬 Cauze și factori de risc
Cauza fundamentală a miotoniei congenitale Thomsen este exclusiv genetică. Aceasta nu este o boală dobândită sau contagioasă.
Cauza genetică: Boala este provocată de mutații (erori în codul genetic) în gena CLCN1. Această genă oferă instrucțiuni pentru fabricarea unei proteine care formează canale de clorură, numite ClC-1, ce se găsesc în membrana celulelor musculare scheletice. Aceste canale joacă un rol crucial în faza de repolarizare (relaxare) a mușchiului. O mutație în gena CLCN1 duce la canale de clorură disfuncționale, care nu permit un flux adecvat de ioni de clor. Rezultatul este o stare de hiperexcitabilitate a membranei musculare, care întârzie relaxarea și produce rigiditatea caracteristică miotoniei [8, 10]. Peste 90% din cazurile de miotonie Thomsen sunt cauzate de astfel de mutații.
Disponibilitatea testării genetice în România
Factori de risc:
- Istoricul familial: Fiind o boală autosomal dominantă, cel mai mare factor de risc este prezența bolii la unul dintre părinți. Un părinte afectat are o probabilitate de 50% de a transmite boala fiecărui copil.
- Expunerea la frig: Deși nu este o cauză, frigul este cel mai important factor de risc pentru declanșarea și agravarea simptomelor la persoanele care au deja predispoziția genetică [1, 2].
⚖️ Tipuri de miotonie congenitală: Thomsen vs. Becker
Miotoniile congenitale non-distrofice sunt clasificate în principal în două tipuri: boala Thomsen și miotonia generalizată Becker. Deși ambele sunt cauzate de mutații în aceeași genă (CLCN1), ele diferă prin modelul de transmitere și severitatea simptomelor [1].
Boala Thomsen (Autosomal Dominantă)
- Transmitere: Autosomal dominantă (o singură genă mutantă este suficientă).
- Debut: Copilărie timpurie sau la naștere.
- Severitate: Formă considerată mai ușoară.
- Slăbiciune musculară: De obicei absentă sau foarte tranzitorie. Forța musculară este normală sau chiar crescută (hipertrofie).
- Progresie: Stabilă, nu se agravează în timp.
Miotonia Becker (Autosomal Recesivă)
- Transmitere: Autosomal recesivă (necesită două copii ale genei mutante, una de la fiecare părinte).
- Debut: Mai târziu în copilărie sau adolescență (între 4 și 18 ani).
- Severitate: Formă mai severă, cu miotonie mai pronunțată.
- Slăbiciune musculară: Adesea prezentă, poate fi tranzitorie sau ușor progresivă în timp, în special după efort. Uneori, pacienții pot dezvolta o slăbiciune permanentă, dar ușoară.
- Progresie: Miotonia poate rămâne stabilă, dar slăbiciunea se poate accentua odată cu vârsta.
🩺 Diagnosticarea miotoniei Thomsen
Diagnosticul bolii Thomsen implică o combinație de evaluare clinică, teste electrofiziologice și confirmare genetică. Procesul este de obicei direct, mai ales dacă există un istoric familial cunoscut.
Metode de diagnostic
-
Evaluare Clinică și Istoric MedicalMedicul neurolog va discuta cu pacientul despre simptome (rigiditate, fenomen de “warm-up”, hipertrofie), momentul debutului, factorii agravanți (frig, repaus) și istoricul familial. Examinarea fizică poate releva hipertrofia musculară și medicul poate testa miotonia direct (ex: rugând pacientul să strângă pumnul sau să închidă ochii strâns).
-
Electromiografia (EMG)Acesta este un test cheie care măsoară activitatea electrică a mușchilor [2]. Un ac subțire (electrod) este introdus în mușchi. La pacienții cu miotonie, EMG-ul înregistrează un sunet caracteristic, descris ca “sunetul unui bombardier în picaj” (dive-bomber sound), care corespunde descărcărilor electrice prelungite ale fibrelor musculare [5]. Această descoperire confirmă prezența miotoniei.
-
Testarea GeneticăDiagnosticul de certitudine se obține prin analiza genetică a unei probe de sânge (tipic 5ml de sânge recoltat pe EDTA) [8]. Secvențierea genei CLCN1 poate identifica mutația specifică responsabilă pentru boală. Acest test nu doar confirmă diagnosticul, ci ajută și la diferențierea clară între forma Thomsen (dominantă) și Becker (recesivă), având implicații pentru consilierea genetică a familiei.
Când să consulți un medic
Este recomandat să consulți un medic, de preferat un neurolog, dacă tu sau copilul tău prezentați oricare dintre următoarele simptome persistente [2]:
- Rigiditate musculară neobișnuită care interferează cu activitățile zilnice.
- Dificultate în a iniția mișcările după repaus.
- Căderi frecvente sau dificultăți de coordonare, în special la copii.
- Creștere vizibilă a masei musculare fără o cauză aparentă (exerciții fizice intense).
- Dacă există un istoric de miotonie în familie.
💊 Tratament și management
Nu există un tratament curativ pentru miotonia congenitală Thomsen, deoarece este o afecțiune genetică. Cu toate acestea, simptomele pot fi gestionate eficient, permițând majorității pacienților să ducă o viață activă și împlinită [2].
Strategii de Management Zilnic
Managementul eficient este la fel de important ca tratamentul medicamentos. Pacienții învață să-și “asculte” corpul și să adopte strategii pentru a minimiza impactul miotoniei. Acestea includ: exerciții de “încălzire” înainte de activități (câteva genuflexiuni înainte de a se ridica), evitarea pe cât posibil a tranzițiilor bruște de la repaus la efort și protejarea împotriva frigului prin purtarea de haine adecvate.
Tratamentul medicamentos: Principalul obiectiv al medicației este reducerea rigidității musculare.
- Blocantele canalelor de sodiu: Acestea sunt cele mai eficiente medicamente. Mexiletina este considerată tratamentul de primă linie și s-a demonstrat că ameliorează semnificativ miotonia la aproximativ 70% dintre pacienți [2]. Alte opțiuni includ lamotrigina sau fenitoina [7]. Aceste medicamente acționează prin stabilizarea membranei celulare musculare, reducând hiperexcitabilitatea care cauzează contracția prelungită.
Tratamentul este personalizat. Nu toți pacienții necesită medicație. Decizia de a începe tratamentul se bazează pe severitatea simptomelor și pe impactul acestora asupra calității vieții. Copiii cu simptome severe pot beneficia de tratament precoce pentru a le facilita participarea la activități școlare și sociale [1, 2]. Tratamentul nu diferă în funcție de sex, dar managementul sarcinii la femeile cu miotonie necesită o monitorizare atentă, deoarece fluctuațiile hormonale și anestezia pot influența simptomele.
🧠 Verifică-ți cunoștințele
Ce genă este responsabilă pentru apariția miotoniei congenitale Thomsen?
⚠️ Complicații posibile
Boala Thomsen este în general considerată o afecțiune benignă, cu risc foarte scăzut de complicații severe sau progresie, spre deosebire de distrofiile miotonice [2]. Totuși, dacă este lăsată netratată sau gestionată necorespunzător, poate afecta calitatea vieții.
- Căderi și accidentări: Rigiditatea bruscă, în special la începerea unei mișcări, poate duce la pierderea echilibrului și la căderi, crescând riscul de fracturi sau alte leziuni.
- Durere cronică: Deși miotonia în sine este adesea descrisă ca nedureroasă, unii pacienți pot dezvolta dureri musculare cronice sau crampe, în special după efort.
- Oboseală: Efortul constant de a depăși rigiditatea musculară poate fi epuizant, ducând la o oboseală generalizată.
- Impact psihosocial: Dificultățile în efectuarea unor sarcini simple și imprevizibilitatea simptomelor pot duce la frustrare, anxietate și izolare socială, în special la copii și adolescenți.
- Complicații anestezice: Pacienții cu miotonie necesită o atenție specială în timpul intervențiilor chirurgicale. Anumite medicamente anestezice (în special blocantele neuromusculare depolarizante precum succinilcolina) pot induce spasme musculare severe și generalizate, punând viața în pericol. Este esențial ca echipa chirurgicală și anestezistul să fie informați despre diagnostic.
❤️ Stil de viață și prevenție
Deoarece boala Thomsen este genetică, nu poate fi prevenită în sensul clasic. Prevenția se concentrează pe evitarea complicațiilor și pe managementul simptomelor. De asemenea, consilierea genetică joacă un rol important pentru familiile afectate.
Remedii și adaptarea stilului de viață:
- Mișcare regulată și încălzire: Activitatea fizică moderată și constantă ajută la menținerea flexibilității. O rutină de “încălzire” cu mișcări ușoare înainte de a se angaja în activități mai intense este crucială [1].
- Evitarea frigului: Purtarea de haine călduroase în straturi, evitarea băilor reci și a expunerii prelungite la aer condiționat poate reduce semnificativ frecvența și severitatea episoadelor de miotonie.
- Masaj și căldură locală: Masajul terapeutic și aplicarea de comprese calde sau băile calde pot ajuta la relaxarea mușchilor încordați [1].
- Terapie fizică (Kinetoterapie): Un kinetoterapeut poate elabora un program personalizat de exerciții de stretching și întărire musculară pentru a îmbunătăți funcționalitatea și a preveni contracturile.
Prevenția la nivel familial:
- Consiliere genetică: Persoanele diagnosticate cu boala Thomsen și partenerii lor pot apela la un consilier genetic. Acesta poate explica riscurile de transmitere a bolii la copii (50% pentru fiecare sarcină) și poate discuta opțiunile disponibile, precum diagnosticul prenatal sau preimplantatoriu.
- Screening familial: Odată ce o mutație CLCN1 este identificată la un membru al familiei, alți membri care prezintă simptome pot fi testați specific pentru acea mutație, permițând un diagnostic rapid și ieftin [8, 10].
❓ Întrebări frecvente (FAQ)
▼
Boala Thomsen debutează de regulă în copilăria timpurie, adesea în primii ani de viață. Părinții pot observa că bebelușul are un plâns însoțit de închiderea prelungită a ochilor sau că are dificultăți la inițierea mișcărilor odată ce începe să meargă [1].
▼
Prognosticul este excelent. Boala Thomsen este o afecțiune benignă, non-progresivă. Nu afectează funcția cardiacă, funcția cognitivă sau speranța de viață. Cu un management adecvat al simptomelor, majoritatea persoanelor trăiesc o viață complet normală și activă [2].
▼
Da. Diagnosticul clinic și prin EMG poate fi realizat de medicii neurologi din România. Testarea genetică pentru confirmarea mutațiilor genei CLCN1 este, de asemenea, disponibilă în laboratoare specializate din țară [8, 10]. Tratamentele medicamentoase, precum mexiletina, sunt de asemenea accesibile, deși pot necesita prescripții speciale, în funcție de disponibilitatea pe piața farmaceutică.
📚 Resurse și informații suplimentare
Surse Medicale de Referință (EN)
National Organization for Rare Disorders (NORD). (n.d.). Myotonia Congenita. rarediseases.org/rare-diseases/myotonia-congenita/
U.S. National Library of Medicine. (2020). Myotonia congenita. MedlinePlus. medlineplus.gov/genetics/condition/myotonia-congenita/
Stunnenberg, B. C., LoRusso, S., & Trivedi, J. R. (2019). CLCN1-Related Myotonia. In M. P. Adam et al. (Eds.), GeneReviews®. University of Washington, Seattle. ncbi.nlm.nih.gov/books/NBK548483/
Muscular Dystrophy Association (MDA). (n.d.). Myotonia Congenita (Thomsen’s and Becker’s). mda.org/disease/myotonia-congenita
The Johns Hopkins University. (n.d.). Myotonia Congenita. hopkinsmedicine.org/health/conditions-and-diseases/myotonia-congenita




