


Sindromul miastenic congenital (CMS) este un grup de afecțiuni genetice rare care afectează comunicarea dintre nervi și mușchi, ducând la slăbiciune și oboseală musculară ce debutează de obicei la naștere sau în copilăria timpurie. Spre deosebire de miastenia gravis, care este o boală autoimună, CMS este cauzat de mutații în peste 35 de gene esențiale pentru funcționarea joncțiunii neuromusculare. Diagnosticul se bazează pe simptome clinice, studii electrofiziologice (EMG) și, în final, pe testare genetică. Deși nu există un tratament curativ, managementul simptomatic, adaptat tipului specific de mutație, poate îmbunătăți semnificativ calitatea vieții pacienților.
- 🧬 Cauză Genetică: CMS este cauzat de mutații moștenite în peste 35 de gene, nu de un atac autoimun.
- 👶 Debut Precoce: Simptomele, precum pleoape căzute (ptoză) și dificultăți de hrănire sau respirație, apar de obicei la naștere sau în primii ani de viață.
- 🔌 Defect de Transmisie: Problema constă într-un defect al „cablajului” la joncțiunea neuromusculară, împiedicând transmiterea corectă a semnalului de la nerv la mușchi.
- 💊 Tratament Personalizat: Managementul depinde de gena afectată; unele forme răspund la inhibitori de acetilcolinesterază, în timp ce altele necesită medicamente precum 3,4-diaminopiridină sau pot fi chiar agravate de anumite tratamente.
- 🌍 Raritate: Este o boală foarte rară, cu o prevalență estimată la sub 1 caz la 100.000 de persoane la nivel global, ceea ce face diagnosticul o provocare.
Cuprins
🧬 Despre Sindromul Miastenic Congenital (CMS)
Sindromul miastenic congenital (CMS) reprezintă un grup heterogen și rar de boli genetice ereditare. Caracteristica principală este slăbiciunea musculară care se agravează la efort (fatigabilitate), manifestată încă de la naștere sau din prima copilărie. Cauza fundamentală este un defect în transmiterea semnalului de la nerv la mușchi la nivelul joncțiunii neuromusculare (JNM) colinergice.
Aceste afecțiuni sunt considerate boli rare. Studiile din Regatul Unit indică o prevalență de aproximativ 9 cazuri la un milion de copii sub 18 ani. La nivel global, au fost raportate puțin peste 1.000 de cazuri, dar numărul real este probabil mai mare din cauza subdiagnosticării. În România, se estimează că există sub 50 de cazuri, diagnosticul și managementul fiind concentrate în centre specializate, precum Institutul Național pentru Sănătatea Mamei și Copilului „Alessandrescu-Rusescu” din București.
Prevalență globală estimată
Gene identificate ca fiind cauzatoare
Dintre cazuri prezintă simptome oculare
Procent risc de moștenire (forma recesivă)
🔌 Ce este CMS? Defectul genetic al joncțiunii neuromusculare
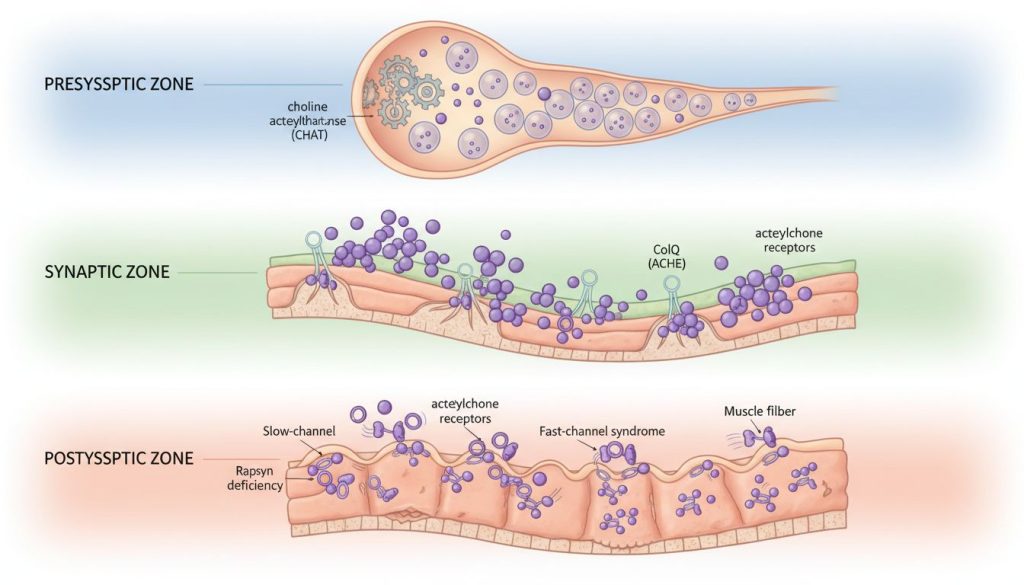
Pentru ca un mușchi să se contracte, un impuls electric trebuie să călătorească de-a lungul unui nerv până la capătul acestuia, unde eliberează un neurotransmițător numit acetilcolină. Acetilcolina traversează un spațiu minuscul, joncțiunea neuromusculară, și se leagă de receptori specifici de pe suprafața mușchiului, declanșând contracția. În CMS, acest proces este defectuos din cauza unor mutații genetice care afectează diverse componente ale joncțiunii.
Este esențial să se facă diferența între CMS și miastenia gravis (MG), o altă boală care provoacă slăbiciune musculară. Deși simptomele pot fi similare, cauzele sunt fundamental diferite.
Sindrom Miastenic Congenital (CMS)
• Cauză: Genetică (mutații ereditare)
• Debut: Congenital (de la naștere) sau infantil
• Mecanism: Defect structural/funcțional al JNM
• Tratament: Simptomatic, specific genei
Miastenia Gravis (MG)
• Cauză: Autoimună (anticorpi atacă JNM)
• Debut: Orice vârstă, frecvent la adulți tineri
• Mecanism: Atac al sistemului imunitar
• Tratament: Imunosupresoare, timectomie
🩺 Simptome
Simptomele CMS sunt variate și depind de gena specifică afectată și de severitatea mutației. Manifestarea centrală este fatigabilitatea musculară, care apare sau se agravează după activitate fizică și se ameliorează cu odihnă. Simptomele pot fi prezente de la naștere (formă congenitală) sau pot apărea în copilărie.
Simptome comune
Majoritatea pacienților (între 80-90% din cazuri) prezintă simptome care afectează mușchii feței, ochilor și gâtului.
- Slăbiciune oculară: Pleoape căzute (ptoză), viziune dublă (diplopie), mișcări oculare limitate (oftalmoplegie).
- Slăbiciune bulbară: Dificultăți de supt, mestecat și înghițit (disfagie), voce nazonată sau slabă (dizartrie).
- Slăbiciune a membrelor: Afectează în special mușchii proximali (umeri, șolduri), ducând la dificultăți la mers, alergat, urcat scări sau ridicarea brațelor.
- Probleme respiratorii: Poate varia de la episoade scurte de oprire a respirației (apnee), în special la sugari, până la insuficiență respiratorie cronică în cazurile severe.
- Hipotonie neonatală: Un tonus muscular foarte scăzut la naștere, copilul părând “moale” (floppy baby).
Manifestări la debut
Simptome rare
Anumite mutații genetice pot duce la manifestări mai puțin obișnuite:
- Atrofie musculară distală: Slăbiciune și pierdere de masă musculară la nivelul mâinilor și picioarelor (asociată cu mutații în gena AGRN).
- Paralizie neonatală: Lipsa completă a mișcării la naștere.
- Comorbidități neurologice: Unele forme foarte rare (ex. mutații în Munc13-1) pot fi asociate cu probleme ale sistemului nervos central, precum microcefalie sau epilepsie.
- Răspuns paradoxal la efort: În unele cazuri, forța musculară poate crește pe parcursul primelor contracții, înainte de a scădea (fenomen de “încălzire”).
🔬 Cauze și factori de risc
CMS este cauzat exclusiv de mutații în genele care codifică proteine esențiale pentru structura și funcția joncțiunii neuromusculare. Până în prezent, au fost identificate peste 35 de astfel de gene. Modul de transmitere genetică este un factor cheie.
Principalul factor de risc este prezența unei mutații cauzatoare în familie. Modurile de moștenire includ:
- Autosomal recesiv (cel mai frecvent): Ambii părinți sunt purtători sănătoși ai unei copii a genei mutante. Există un risc de 25% la fiecare sarcină ca un copil să moștenească ambele copii mutante și să dezvolte boala. 50% dintre copii vor fi purtători sănătoși, iar 25% vor fi neafectați.
- Autosomal dominant: O singură copie a genei mutante, moștenită de la un părinte afectat, este suficientă pentru a cauza boala. Riscul pentru fiecare copil este de 50%.
- Mutații *de novo*: Ocazional, mutația poate apărea spontan la copil, fără a fi prezentă la niciunul dintre părinți. Aceste cazuri sunt mai rare.
Moștenire Autosomal Recesivă (majoritatea cazurilor)
- Părinții sunt de obicei asimptomatici (doar purtători).
- Boala nu apare în fiecare generație a familiei.
Considerații de Risc
- Risc de 25% ca fiecare copil să fie afectat.
- Risc de 50% ca fiecare copil să fie purtător.
- Consangvinitatea (înrudirea părinților) crește riscul.
🧩 Tipuri de CMS
CMS este clasificat în funcție de localizarea defectului la nivelul joncțiunii neuromusculare. Această clasificare este crucială, deoarece tratamentul variază semnificativ între tipuri.
| Tip CMS | Localizarea Defectului | Gene Comune | Prevalență Aproximativă | Mecanism Defectuos |
|---|---|---|---|---|
| Postsinaptic | Membrana musculară | CHRNE, CHRNA1, CHRND, CHRGB1 (subunități AChR), DOK7, RAPSN, MUSK | ~50-70% (cel mai comun) | Număr redus de receptori de acetilcolină (AChR) sau funcționarea anormală a acestora (ex: canale care se închid prea repede – “fast-channel” sau prea lent – “slow-channel”). |
| Presinaptic | Terminalul nervos | CHAT, SYT2, SNAP25, MUNC13-1 | ~15-20% | Sinteză sau eliberare deficitară a acetilcolinei din terminația nervoasă. |
| Sinaptic | Spațiul sinaptic (între nerv și mușchi) | COLQ, LAMB2, AGRN, LRP4 | ~10-15% | Defect al enzimei care descompune acetilcolina (acetilcolinesterază) sau al proteinelor structurale care ancorează JNM. |
🧑⚕️ Diagnosticarea CMS
Diagnosticul CMS este un proces complex care implică excluderea altor afecțiuni și combinarea datelor clinice, electrofiziologice și genetice. De obicei, diagnosticul este suspectat la sugari sau copii mici cu hipotonie și slăbiciune fluctuantă.
-
Evaluare ClinicăMedicul neurolog pediatru evaluează istoricul medical, simptomele (ptoză, fatigabilitate) și efectuează un examen fizic detaliat.
-
Testare Electrofiziologică (EMG/RNS)Se realizează electromiografia (EMG) cu stimulare nervoasă repetitivă (RNS) la o frecvență joasă (2-3 Hz). Un răspuns caracteristic este scăderea progresivă (decrement) a amplitudinii potențialului de acțiune muscular compus (CMAP), indicând un defect de transmisie neuromusculară.
-
Testare GeneticăConfirmarea finală a diagnosticului se face prin analiza ADN dintr-o probă de sânge. Se utilizează paneluri de secvențiere de nouă generație (NGS) care analizează simultan toate genele cunoscute a fi asociate cu CMS, pentru a identifica mutația cauzatoare.
În plus, se efectuează teste de sânge pentru a exclude miastenia gravis autoimună prin căutarea anticorpilor specifici (anti-AChR, anti-MuSK).
🚨 Când să consulți un medic
Părinții ar trebui să solicite o evaluare medicală specializată dacă observă la copilul lor oricare dintre următoarele semne, în special dacă acestea sunt persistente sau se agravează la oboseală:
- Pleoape vizibil căzute (ptoză), uni- sau bilaterală.
- Dificultăți persistente la hrănire (supt slab, oboseală în timpul mesei, înecare frecventă).
- Plâns slab, răgușit.
- Tonus muscular redus (“copil moale”).
- Episoade de oprire a respirației (apnee), în special în timpul somnului sau hrănirii.
- Slăbiciune generalizată sau oboseală inexplicabilă după efort fizic normal pentru vârsta sa.
- Întârziere în dezvoltarea motorie (nu-și ține capul, nu se rostogolește, nu merge la vârsta potrivită).
Sfat pentru părinți
Filmați episoadele de slăbiciune, ptoză sau dificultăți respiratorii pentru a le arăta medicului. Un jurnal al simptomelor, notând când apar și ce pare să le declanșeze (efort, febră, infecții), poate fi extrem de util în procesul de diagnostic.
💊 Tratament
În prezent, nu există un tratament care să vindece CMS. Cu toate acestea, există multiple opțiuni de tratament simptomatic care pot controla eficient slăbiciunea musculară și pot îmbunătăți semnificativ calitatea vieții. Tratamentul este strict personalizat în funcție de gena afectată.
- Inhibitori de acetilcolinesterază (ex: Piridostigmină): Aceștia cresc cantitatea de acetilcolină disponibilă în joncțiune prin încetinirea descompunerii sale. Sunt eficienți în multe forme postsinaptice (ex: deficit de receptori), dar pot agrava alte forme, cum ar fi CMS-ul de tip “slow-channel” sau cel cauzat de deficitul de ColQ.
- 3,4-Diaminopiridină (3,4-DAP): Acest medicament crește eliberarea de acetilcolină din terminația nervoasă. Este tratamentul de elecție pentru majoritatea formelor presinaptice (ex: mutații în gena CHAT).
- Salicilați (Aspirină) sau Fluoxetină: Utilizate în tratamentul formelor “slow-channel” (ex: anumite mutații ale receptorilor AChR), unde ajută la scurtarea timpului în care canalele ionice rămân deschise, prevenind acumularea excesivă de calciu în celula musculară.
- Efedrină sau Albuterol (Salbutamol): Aceste medicamente β2-adrenergice s-au dovedit a fi eficiente în anumite tipuri de CMS, cum ar fi cele cauzate de mutații în genele DOK7 sau AGRN, unde par să ajute la stabilizarea și organizarea joncțiunii neuromusculare.
În peste 80% din cazuri, odată ce tipul genetic este identificat corect, se poate găsi un tratament care aduce beneficii vizibile.
🧠 Verifică-ți cunoștințele
Care este principala diferență între sindromul miastenic congenital (CMS) și miastenia gravis (MG)?
❤️🩹 Managementul CMS
Managementul pe termen lung se concentrează pe monitorizarea funcțiilor vitale, terapie de susținere și adaptarea stilului de viață.
Stil de viață și remedii la domiciliu
- Managementul energiei: Pacienții trebuie să învețe să își dozeze efortul și să alterneze perioadele de activitate cu cele de odihnă programată pentru a preveni epuizarea.
- Fizioterapie și kinetoterapie: Programele de exerciții moderate ajută la menținerea forței musculare, a mobilității articulare și la prevenirea atrofiilor.
- Evitarea factorilor declanșatori: Infecțiile, febra, stresul și anumite medicamente (precum unele antibiotice, relaxante musculare sau betablocante) pot agrava slăbiciunea și trebuie gestionate cu prudență, sub supraveghere medicală.
- Suport nutrițional: O dietă echilibrată, bogată în calorii și proteine, este esențială, în special pentru copiii cu dificultăți de hrănire. Uneori poate fi necesară hrănirea prin gastrostomă.
Diferențe de tratament în funcție de sex și vârstă
Boala afectează în mod egal ambele sexe. Nu există diferențe de tratament bazate pe sex. Cu toate acestea, dozele medicamentelor trebuie ajustate în funcție de vârstă și greutate. Nou-născuții cu forme severe pot necesita îngrijiri intensive, inclusiv ventilație mecanică. Pe măsură ce copilul crește, dozele de medicamente (precum piridostigmina) trebuie crescute corespunzător pentru a menține eficacitatea.
⚠️ Complicații posibile
Cea mai gravă complicație a CMS este insuficiența respiratorie. Crizele miastenice, declanșate de infecții sau alți factori de stres, pot duce la slăbiciune severă a mușchilor respiratori, necesitând ventilație de urgență.
- Crize respiratorii: Pot fi letale, în special în perioada neonatală, unde mortalitatea poate atinge 10-20% în cazurile severe. Aproximativ 20% dintre pacienții cu forme grave necesită ventilație non-invazivă sau invazivă pe termen lung.
- Probleme de hrănire: Disfagia poate duce la malnutriție, deshidratare și pneumonie de aspirație.
- Complicații ortopedice: Slăbiciunea cronică poate cauza deformări ale coloanei vertebrale (scolioză), contracturi articulare și probleme de mobilitate.
- Atrofie musculară: Lipsa utilizării normale a mușchilor poate duce la pierderea masei musculare în timp.
🛡️ Prevenție
Deoarece CMS este o boală genetică, nu poate fi prevenită în sensul clasic. Singura metodă de prevenție este cea la nivel de planificare familială.
- Consiliere genetică: Familiile în care există un caz de CMS sau care au un istoric familial pot beneficia de consiliere genetică. Aceasta ajută la înțelegerea riscurilor de transmitere a bolii.
- Testare prenatală: Dacă mutația cauzatoare a fost identificată la un membru al familiei, se poate efectua testarea prenatală (prin amniocenteză sau biopsia vilozităților coriale) pentru a determina dacă fătul a moștenit mutația.
- Testarea purtătorilor: Frații neafectați ai unui pacient cu CMS în formă recesivă pot fi testați pentru a vedea dacă sunt purtători ai genei mutante.
❓ Întrebări frecvente despre CMS
▼
CMS este considerată o boală foarte rară. Estimările variază, dar prevalența la nivel mondial este sub 1 caz la 100.000 de persoane. Anumite studii regionale, precum cele din Marea Britanie, arată o prevalență de aproximativ 9.2 la 1 milion de copii.
▼
Cel mai frecvent, CMS se moștenește într-un mod autosomal recesiv, ceea ce înseamnă că ambii părinți trebuie să fie purtători ai unei gene defecte pentru ca un copil să aibă un risc de 25% de a dezvolta boala. Mai rar, se poate transmite autosomal dominant (risc de 50% de la un părinte afectat).
▼
Nu, în prezent nu există un tratament curativ. Totuși, tratamentele simptomatice sunt adesea foarte eficiente și pot îmbunătăți dramatic forța musculară și calitatea vieții. Cercetările sunt în curs de desfășurare pentru terapii genice și alte abordări inovatoare.
▼
Diferența fundamentală este cauza. CMS este o boală genetică, cauzată de o mutație moștenită care afectează funcționarea joncțiunii neuromusculare. MG este o boală autoimună, în care propriul sistem imunitar al organismului produce anticorpi care atacă și distrug componente ale aceleiași joncțiuni.
📚 Referințe / Surse
Abicht, A., & Müller, J. S. (2017). Congenital Myasthenic Syndromes or Inherited Disorders of the Neuromuscular Junction. GeneReviews®. University of Washington, Seattle. pmc.ncbi.nlm.nih.gov/articles/PMC5701762/
Cleveland Clinic. (n.d.). Congenital Myasthenic Syndrome (CMS). my.clevelandclinic.org/health/diseases/congenital-myasthenic-syndrome
Muscular Dystrophy Association. (n.d.). Understanding Congenital Myasthenic Syndrome: Causes and Treatments. MDA Quest Magazine. mdaquest.org/understanding-congenital-myasthenic-syndrome-causes-and-treatments/
Oh, S. J. (2023). Congenital myasthenic syndromes. MedLink Neurology. www.medlink.com/articles/congenital-myasthenic-syndromes
Mayo Clinic. (n.d.). Congenital myasthenic syndromes – Symptoms and causes. www.mayoclinic.org/diseases-conditions/congenital-myasthenic-syndrome/symptoms-causes/syc-20354754
Muscular Dystrophy Association. (n.d.). Congenital Myasthenic Syndromes (CMS). www.mda.org/disease/congenital-myasthenic-syndromes




