


Articolul explorează în detaliu tipurile de CJD (sporadică, ereditară și iatrogenă), simptomele devastatoare cu evoluție rapidă, de la modificări de personalitate la demență severă și mișcări involuntare. De asemenea, sunt abordate metodele complexe de diagnostic, managementul simptomatic (în absența unui tratament curativ), prognosticul sumbru și măsurile esențiale de prevenție, în special în context medical.
- 🧠 Cauză Unică: CJD este cauzată de prioni, proteine anormale care distrug țesutul cerebral, nu de viruși sau bacterii.
- 📈 Progresie Rapidă: Spre deosebire de alte forme de demență, CJD progresează extrem de rapid, ducând la deteriorare severă în câteva luni.
- 🚫 Fără Tratament: În prezent, nu există un leac pentru CJD; tratamentul este paliativ, concentrându-se pe ameliorarea simptomelor.
- 🌍 Raritate: Este o boală foarte rară, cu o incidență de aproximativ 1-2 cazuri la un milion de persoane pe an la nivel global.
- 🔬 Diagnostic Definitiv: Singura modalitate de a confirma 100% diagnosticul este prin analiza țesutului cerebral, efectuată de obicei post-mortem.
Cuprins
🧠 Despre Boala Creutzfeldt-Jakob (Ce este?)
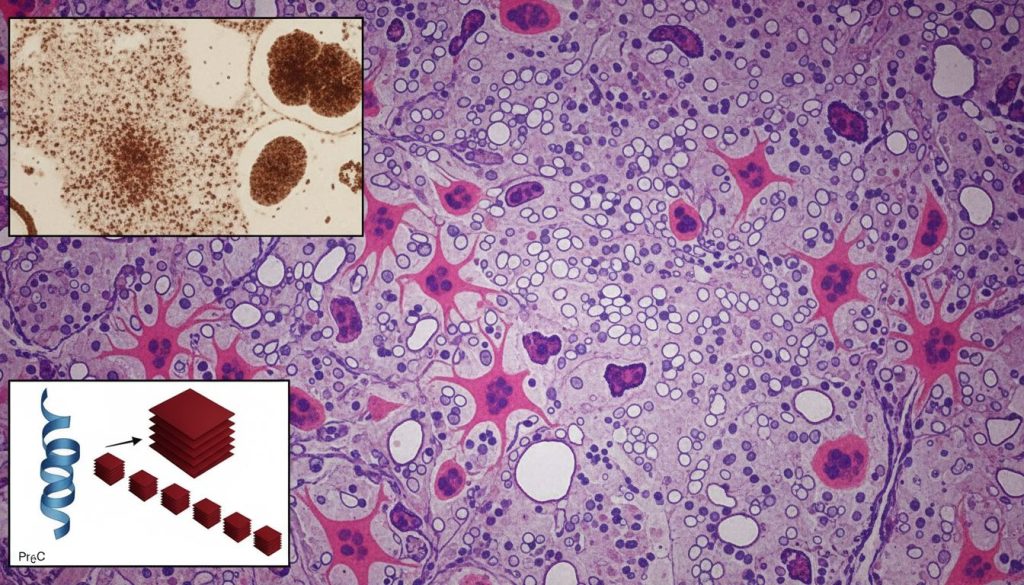
Boala Creutzfeldt-Jakob (CJD) este o afecțiune neurologică degenerativă, rară și invariabil fatală, care face parte dintr-o familie de boli cunoscute sub numele de encefalopatii spongiforme transmisibile (EST). Este cea mai comună formă de boală prionică la om. Numele “spongiform” provine de la aspectul post-mortem al creierului afectat, care dezvoltă găuri microscopice, semănând cu un burete. Această deteriorare este rezultatul direct al distrugerii celulelor nervoase (neuronilor).
Agentul cauzal nu este un virus, o bacterie sau o ciupercă, ci un tip special de proteină numit prion. Prionii sunt agenți infecțioși nonconvenționali, lipsiți de material genetic (ADN sau ARN), care sunt de fapt forme pliate anormal (misfoldate) ale unei proteine celulare normale, cunoscută ca proteina prionică (PrPC).
Mecanismul bolii este devastator: proteinele prionice anormale (notate PrPSc) se acumulează în creier și sunt rezistente la mecanismele de curățare ale celulei. Această acumulare este toxică pentru neuroni, ducând la disfuncția și moartea lor. Rezultatul este un declin cognitiv și motor extrem de rapid, mult mai accelerat decât în alte boli neurodegenerative, cum ar fi Alzheimer.
🧬 Tipuri de CJD
Boala Creutzfeldt-Jakob se clasifică în trei subtipuri principale, în funcție de modul de apariție.
CJD Sporadică
CJD Ereditară
CJD Iatrogenă / Dobândită
CJD Variantă (vCJD)
CJD Sporadică
Aceasta este cea mai comună formă, reprezentând aproximativ 85% din toate cazurile. “Sporadic” înseamnă că boala apare aleatoriu, fără o cauză cunoscută sau un factor de risc identificabil. Teoria principală este că o proteină PrPC normală suferă o modificare spontană, transformându-se în forma prionică PrPSc, care apoi inițiază reacția în lanț. Afectează de obicei persoanele cu vârsta cuprinsă între 60 și 65 de ani.
CJD Ereditară (Familială)
Reprezintă între 5% și 10% din cazuri și este cauzată de o mutație genetică moștenită în gena care codifică proteina prionică (gena PRNP). Această mutație face ca proteina să fie mult mai susceptibilă la plierea greșită. Dacă o persoană moștenește gena mutantă de la un părinte, riscul de a dezvolta boala este de 50%. În această categorie intră și alte boli prionice extrem de rare, precum sindromul Gerstmann-Sträussler-Scheinker (GSS) și insomnia fatală familială (IFF). Debutul poate fi la o vârstă mai tânără comparativ cu forma sporadică.
CJD Iatrogenă (Dobândită)
Aceasta este cea mai rară formă (sub 1% din cazuri) și apare prin expunerea la țesut cerebral sau spinal contaminat în timpul unei proceduri medicale. Sursele istorice de transmitere includ:
- Transplanturi de țesut: Utilizarea de grefe de dura mater (membrana care acoperă creierul) sau cornee de la donatori nediagnosticați cu CJD.
- Hormon de creștere uman: Administrarea de hormon de creștere derivat din glandele pituitare (hipofiză) ale cadavrelor umane, o practică abandonată în anii ’80 în favoarea hormonilor sintetici.
- Instrumente neurochirurgicale contaminate: Prionii sunt extrem de rezistenți la metodele standard de sterilizare.
O formă distinctă de CJD dobândită este CJD variantă (vCJD), legată de consumul de carne de vită provenită de la animale infectate cu encefalopatie spongiformă bovină (ESB), cunoscută popular ca “boala vacii nebune”.
📈 Simptome și Evoluție
Semnul distinctiv al CJD este progresia extrem de rapidă a simptomelor. În timp ce alte demențe, precum Alzheimer, pot evolua pe parcursul a mulți ani, declinul în CJD se produce în decurs de săptămâni sau luni.
Simptome comune (cu progresie rapidă)
- Tulburări cognitive: Pierderea rapidă a memoriei, dificultăți de gândire, judecată afectată, confuzie și dezorientare.
- Modificări de personalitate: Depresie, anxietate, agitație, apatie sau iritabilitate bruscă.
- Probleme motorii:
- Mioclonii: Spasme musculare bruște, asemănătoare unor șocuri electrice. Acesta este un simptom caracteristic.
- Ataxie: Pierderea coordonării, mers instabil, tremurături și dificultăți în efectuarea mișcărilor precise.
- Rigiditate musculară: Mușchii devin înțepeniți.
- Tulburări vizuale: Vedere încețoșată, halucinații vizuale sau chiar orbire.
- Dificultăți de limbaj și înghițire (disfagie): Vorbire neclară, greutate în a forma cuvinte și risc de înec.
- Insomnie severă.
Evoluția bolii
Progresia CJD este implacabilă și poate fi vizualizată ca o succesiune rapidă de etape.
-
Stadiul Inițial (Săptămâni): Simptome vagi, precum anxietate, depresie, mici probleme de memorie și coordonare. Pot fi ușor confundate cu alte afecțiuni.
-
Stadiul de Progresie (Luni): Declinul cognitiv devine evident și rapid. Apar miocloniile, ataxia se agravează, iar pacientul devine dependent de ajutor pentru activitățile zilnice.
-
Stadiul Final (Luni): Pacientul pierde complet funcțiile mentale și fizice. Intră într-o stare de mutism akinetic (nu mai poate vorbi sau mișca voluntar), ajungând în cele din urmă în comă. Decesul survine adesea din cauza complicațiilor, precum pneumonia de aspirație sau alte infecții.
🧪 Cauze și Factori de Risc
Cauza fundamentală a CJD este transformarea proteinei PrPC în forma sa patologică PrPSc. Factorii care declanșează acest proces definesc tipul bolii și riscurile asociate.
Factori de Risc Cunoscuți
- Vârsta: Riscul pentru CJD sporadică crește semnificativ cu vârsta, majoritatea cazurilor apărând la persoane de peste 60 de ani.
- Genetică: Prezența unei mutații în gena PRNP este un factor de risc absolut pentru CJD ereditară. Istoricul familial de CJD este un semnal de alarmă.
- Expunere Iatrogenă: Persoanele care au suferit anumite proceduri neurochirurgicale, au primit grefe de dura mater sau au fost tratate cu hormon de creștere uman înainte de 1985 prezintă un risc crescut.
Factori Fără Risc Dovedit
- Contact Ocazional: CJD nu se transmite prin contact social obișnuit, atingere, tuse, strănut sau contact sexual. Îngrijirea unui pacient cu CJD nu prezintă risc dacă se evită contactul cu țesutul cerebral sau lichidul cefalorahidian.
- Transfuzii de Sânge: Pentru CJD clasică (sporadică, ereditară, iatrogenă), nu există dovezi clare de transmitere prin sânge, deși teoretic riscul nu este zero. Pentru vCJD, transmiterea prin sânge a fost documentată.
Atenție la Riscul Ocupațional
Personalul medical, în special neurochirurgii, patologii și tehnicienii de laborator care manipulează țesuturi umane din sistemul nervos central, prezintă un risc teoretic mai mare. Respectarea strictă a protocoalelor de siguranță și decontaminare este esențială.
🩺 Diagnosticare
Diagnosticarea CJD în timpul vieții este dificilă, deoarece multe simptome seamănă cu cele ale altor tulburări neurologice mai comune. Diagnosticul definitiv poate fi pus doar prin biopsie cerebrală sau autopsie. Cu toate acestea, o combinație de teste poate oferi un diagnostic foarte probabil.
-
Evaluare Clinică și AnamnezăMedicul neurolog evaluează simptomele caracteristice (demență rapidă, mioclonii, ataxie) și exclude alte afecțiuni, precum Alzheimer, encefalită, tumori cerebrale sau accidente vasculare cerebrale.
-
Investigații Imagistice și ElectriceImagistica prin Rezonanță Magnetică (RMN): Poate evidenția modificări specifice în anumite zone ale creierului (ex. cortex, ganglioni bazali) care sunt sugestive pentru CJD.
Electroencefalograma (EEG): Poate înregistra un model specific de activitate electrică anormală (complexe periodice trifazice), prezent în aproximativ două treimi din cazurile de CJD sporadică. -
Analize de Laborator SpecificePuncția lombară: Analiza lichidului cefalorahidian (LCR) este crucială. Teste moderne, precum RT-QuIC (Real-Time Quaking-Induced Conversion), pot detecta prezența proteinelor prionice anormale cu o specificitate și sensibilitate foarte înalte. Prezența proteinei 14-3-3 este un alt marker, deși mai puțin specific.
Testare genetică: Analiza sângelui poate identifica mutațiile genei PRNP în cazurile suspectate de CJD ereditară.
Creier Sănătos (Conceptual)
• Structură neuronală intactă
• Proteine PrPC normale, funcționale
• Funcții cognitive și motorii normale
Creier Afectat de CJD (Conceptual)
• Apariția de vacuole (aspect spongiform)
• Acumulare de prioni PrPSc toxici
• Demență rapidă și disfuncții motorii severe
💊 Tratament și Management
În prezent, nu există niciun tratament care să vindece, să oprească sau măcar să încetinească progresia bolii Creutzfeldt-Jakob. Boala este întotdeauna fatală. Managementul se concentrează exclusiv pe îngrijirea paliativă și pe ameliorarea simptomelor pentru a oferi pacientului un grad cât mai mare de confort.
Managementul Simptomatic
- Miocloniile (spasmele musculare): Se pot utiliza medicamente anticonvulsivante sau miorelaxante, precum clonazepam sau valproat de sodiu.
- Durerea: Se administrează analgezice, inclusiv opioide în stadiile avansate, pentru a gestiona durerea cauzată de rigiditatea musculară și imobilitate.
- Simptomele psihice (agitație, anxietate): Antidepresivele și sedativele pot ajuta la calmarea pacientului și la îmbunătățirea stării de spirit.
- Îngrijirea generală: Pe măsură ce boala progresează, pacientul va necesita asistență completă pentru hrănire (adesea prin sondă nazogastrică), igienă și repoziționare pentru a preveni escarele.
Studiile clinice sunt în desfășurare în centre specializate, testând diverse molecule care ar putea inhiba conversia proteinelor prionice sau ar putea facilita eliminarea acestora. Participarea la astfel de studii poate fi o opțiune pentru anumiți pacienți, dar nu oferă nicio garanție de succes.
⌛ Prognostic
Prognosticul pentru pacienții cu CJD este extrem de sumbru. Este una dintre cele mai rapid progresive boli neurodegenerative.
Factorii Prognosticului în CJD
95% (Extrem de rapidă)
~10%
0% (Inexistentă)
100% (Inevitabil fatală)
Majoritatea pacienților (aproximativ 90%) decedează în decurs de un an de la apariția primelor simptome. Durata medie de supraviețuire este de aproximativ 4-6 luni. Sprijinul familiei și îngrijirea în centre de tip hospice sunt esențiale pentru a asigura demnitatea și confortul pacientului în stadiile terminale.
🛡️ Prevenție
Nu există o metodă de a preveni CJD sporadică sau ereditară. Măsurile de prevenție se concentrează în totalitate pe stoparea transmiterii formei iatrogene și a celei variante (vCJD).
- Sterilizarea instrumentelor medicale: Spitalele utilizează protocoale stricte de decontaminare pentru instrumentele chirurgicale folosite în proceduri care implică sistemul nervos. Acestea includ tratamente chimice puternice (ex. hidroxid de sodiu) combinate cu autoclavare la temperaturi și presiuni ridicate.
- Instrumente de unică folosință: Ori de câte ori este posibil, se folosesc instrumente de unică folosință în neurochirurgie.
- Restricții la donarea de sânge și țesuturi: Persoanele cu risc de CJD (istoric familial, foști beneficiari de grefe de dura mater etc.) nu sunt eligibile pentru a dona sânge, organe sau țesuturi.
- Hormoni sintetici: Utilizarea exclusivă a hormonului de creștere sintetic a eliminat riscul de transmitere pe această cale.
- Siguranța alimentară: Măsuri stricte de control în industria cărnii de vită (ex. interzicerea hrănirii animalelor cu făinuri de origine animală) au redus drastic incidența vCJD.
🧠 Test de Cunoștințe
Care este agentul patogen responsabil pentru declanșarea bolii Creutzfeldt-Jakob?
ℹ️ Informații Suplimentare Importante
- Raritate: CJD este o boală extrem de rară, cu o incidență globală estimată la aproximativ 1-2 cazuri la un milion de locuitori pe an.
- Demografie: Nu există diferențe semnificative în incidență sau tratament între sexe. Majoritatea cazurilor sporadice apar la persoane în vârstă.
- Sprijin pentru familie: Diagnosticul de CJD este devastator pentru familie. Resursele de consiliere psihologică și grupurile de sprijin sunt cruciale pentru a ajuta aparținătorii să facă față progresiei rapide a bolii și pierderii iminente.
❓ Întrebări Frecvente
▼
Nu în sensul convențional. CJD nu se transmite prin aer, atingere, tuse, strănut sau contact social obișnuit. Riscul de transmitere există doar prin contact direct cu țesuturi infectate ale sistemului nervos central (creier, măduva spinării) sau, în cazuri foarte rare și specifice, prin proceduri medicale invazive cu instrumente contaminate sau transplant de țesuturi.
▼
Deși ambele sunt boli neurodegenerative care cauzează demență, există diferențe majore. Cea mai importantă este viteza de progresie: Alzheimer evoluează lent, pe parcursul a 5-15 ani, în timp ce CJD duce la deces în mai puțin de un an. În plus, simptomele motorii precum miocloniile sunt caracteristice CJD, dar rare în Alzheimer. Cauzele sunt, de asemenea, diferite: prioni în CJD versus plăci de beta_amiloid și acumulări de proteină tau în Alzheimer.
▼
Forma de CJD legată de consumul de carne contaminată se numește CJD variantă (vCJD) și este extrem de rară. Datorită măsurilor drastice de siguranță alimentară implementate la nivel mondial după epidemia din Marea Britanie din anii ’90, riscul de a contracta vCJD din consumul de carne de vită, în prezent, este considerat neglijabil în majoritatea țărilor.
📚 Referințe / Surse
Informațiile prezentate în acest articol se bazează pe date de la organizații medicale de renume. Mai jos sunt câteva dintre sursele utilizate:
- Mayo Clinic. (n.d.). Creutzfeldt-Jakob disease – Symptoms & causes. mayoclinic.org/diseases-conditions/creutzfeldt-jakob-disease/symptoms-causes/syc-20371226
- Cleveland Clinic. (n.d.). Creutzfeldt-Jakob Disease (CJD): Symptoms & Treatment. my.clevelandclinic.org/health/diseases/6001-creutzfeldt-jakob-disease
- Centers for Disease Control and Prevention (CDC). (n.d.). Classic Creutzfeldt-Jakob Disease | Classic CJD. cdc.gov/prions/cjd/about.html
- Alzheimer’s Association. (n.d.). Creutzfeldt-Jakob Disease (CJD) | Symptoms & Treatments. alz.org/alzheimers-dementia/what-is-dementia/types-of-dementia/creutzfeldt-jakob-disease
- NHS. (n.d.). Treatment – Creutzfeldt-Jakob disease. www.nhs.uk/conditions/creutzfeldt-jakob-disease-cjd/treatment/
- UCSF Health. (n.d.). Creutzfeldt-Jakob Disease | Conditions. www.ucsfhealth.org/conditions/creutzfeldt-jakob-disease




