
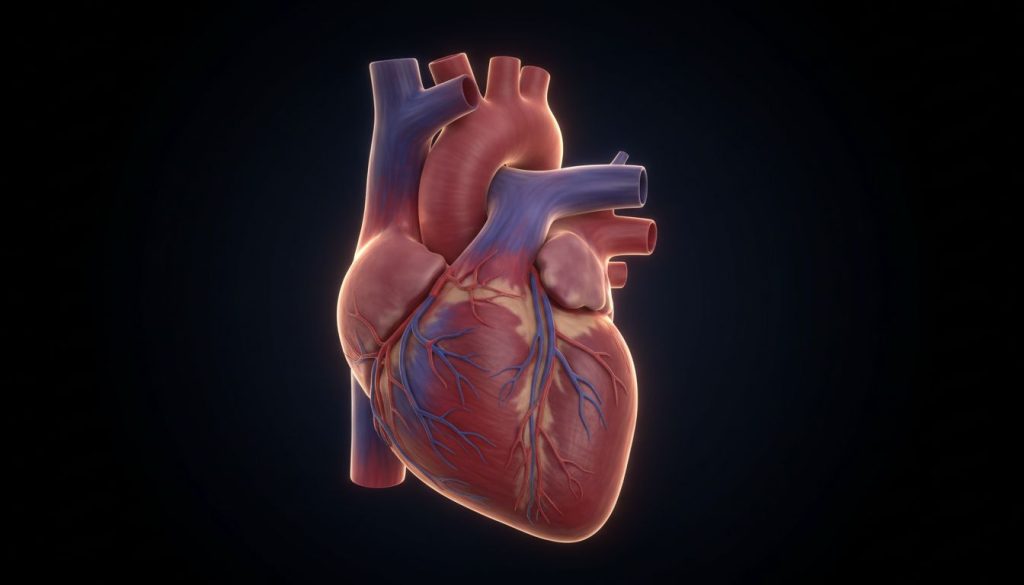
Trunchiul arterial comun, sau truncus arteriosus, este o malformație cardiacă congenitală rară, dar gravă, în care din inimă izvorăște un singur vas de sânge mare, în loc de două (aorta și artera pulmonară). Această anomalie duce la amestecul sângelui oxigenat cu cel neoxigenat, provocând cianoză (colorație albăstruie a pielii) și insuficiență cardiacă severă la scurt timp după naștere. Fără o intervenție chirurgicală corectivă în primele săptămâni sau luni de viață, prognosticul este extrem de nefavorabil.
Acest articol detaliat explorează anatomia specifică a trunchiului arterial comun, simptomele sale caracteristice la nou-născuți, cauzele și factorii de risc asociați, metodele moderne de diagnostic prenatal și postnatal, precum și opțiunile de tratament chirurgical care pot schimba radical destinul acestor copii. De asemenea, vom discuta despre complicațiile posibile și managementul pe termen lung, esențial pentru o calitate bună a vieții.
- 🧬 Malformație complexă: Un singur trunchi vascular înlocuiește aorta și artera pulmonară, fiind aproape întotdeauna asociat cu un defect septal ventricular (DSV).
- 🔵 Simptome alarmante: Cianoză, dificultăți de respirație și alimentație, oboseală, semne clare de insuficiență cardiacă la nou-născut.
- 🩺 Diagnostic esențial: Ecocardiografia fetală permite depistarea înainte de naștere, iar cea postnatală confirmă diagnosticul și detaliile anatomice.
- ❤️🩹 Chirurgie salvatoare: Intervenția chirurgicală este unica soluție și se realizează de urgență în primele luni de viață pentru a repara structura inimii.
- 📈 Prognostic pe termen lung: Cu o corecție chirurgicală reușită, supraviețuirea este semnificativ îmbunătățită, dar necesită monitorizare cardiologică pe tot parcursul vieții.
Cuprins
🧬 Ce este Trunchiul Arterial Comun?
Trunchiul arterial comun (TAC), cunoscut medical ca truncus arteriosus, este o malformație cardiacă congenitală (prezentă de la naștere) în care, în timpul dezvoltării embrionare, vasul unic de sânge care pleacă de la inimă nu reușește să se dividă în cele două artere principale: aorta și artera pulmonară. Drept urmare, un singur trunchi arterial mare izvorăște din ambele ventricule ale inimii, deasupra unui defect septal ventricular (DSV) – o comunicare anormală între cele două camere inferioare ale inimii.
Această condiție face parte din grupul malformațiilor cardiace congenitale cianogene, deoarece permite amestecarea sângelui bogat în oxigen (din partea stângă a inimii) cu cel sărac în oxigen (din partea dreaptă). Acest amestec este apoi pompat atât spre plămâni, cât și spre restul corpului, ducând la o oxigenare ineficientă a țesuturilor și la simptome severe încă din primele zile de viață. Incidența sa este rară, reprezentând aproximativ 1% din totalul malformațiilor cardiace congenitale.
❤️🩹 Fiziologie și Anatomie Afectată
Pentru a înțelege gravitatea trunchiului arterial comun, este esențial să comparăm această anomalie cu anatomia normală a inimii.
Diferența față de anatomia normală
Anatomie Normală
• Inima are 4 camere și 2 circuite separate.
• Ventriculul drept pompează sânge neoxigenat în artera pulmonară spre plămâni.
• Ventriculul stâng pompează sânge oxigenat în aortă spre corp.
• Valva aortică și valva pulmonară sunt separate.
Anatomie cu Truncus Arteriosus
• Există un trunchi arterial unic care pleacă din ambele ventricule.
• Un defect septal ventricular (DSV) mare permite amestecul sângelui.
• O singură valvă, valva truncală, controlează ieșirea sângelui din inimă.
• Sângele amestecat este trimis simultan și spre plămâni, și spre corp.
Consecințele acestei anatomii anormale sunt severe:
- Supraîncărcarea plămânilor: O cantitate prea mare de sânge este pompată către plămâni sub presiune înaltă, ducând la congestie pulmonară și insuficiență respiratorie.
- Efort cardiac crescut: Inima trebuie să pompeze mult mai puternic pentru a susține ambele circulații, ceea ce duce rapid la insuficiență cardiacă.
- Hipoxemie și cianoză: Deoarece sângele care ajunge în corp este un amestec, nivelul de oxigen este scăzut, provocând cianoza (colorație albăstruie a pielii, buzelor și unghiilor).
Clasificarea Trunchiului Arterial Comun
Există mai multe sisteme de clasificare, cel mai frecvent utilizat fiind sistemul Collett și Edwards, care împarte TAC în funcție de originea arterelor pulmonare din trunchiul comun:
- Tip I (cel mai comun): Un trunchi pulmonar principal scurt se desprinde din trunchiul arterial comun și apoi se bifurcă în arterele pulmonare stângă și dreaptă.
- Tip II: Arterele pulmonare stângă și dreaptă apar separat, dar apropiat, de pe peretele posterior al trunchiului comun.
- Tip III: Arterele pulmonare stângă și dreaptă apar separat și la distanță una de cealaltă, de pe pereții laterali ai trunchiului comun.
- Tip IV (istoric): Această formă, în care plămânii sunt irigați de vase colaterale din aorta descendentă, este acum clasificată mai degrabă ca o formă severă de atrezie pulmonară cu defect septal ventricular, și nu ca un tip de truncus arteriosus.
🩺 Simptome și Prezentare Clinică
Simptomele trunchiului arterial comun apar de obicei în primele ore sau zile de viață și sunt adesea dramatice. Nou-născuții pot părea relativ bine imediat după naștere, dar starea lor se deteriorează rapid pe măsură ce rezistența vasculară pulmonară scade.
Semne de alarmă la nou-născut
La examenul fizic, medicul poate detecta un suflu cardiac puternic, un puls periferic accentuat și semne de insuficiență cardiacă, cum ar fi hepatomegalia (ficat mărit). Netratată, această condiție este fatală, 85% dintre pacienți decedând în primul an de viață din cauza insuficienței cardiace severe și a hipoxiei cronice.
🔬 Cauze și Factori de Risc
Cauza exactă a trunchiului arterial comun, la fel ca în cazul multor malformații cardiace congenitale, este adesea necunoscută. Se consideră a fi rezultatul unei combinații de factori genetici și de mediu care intervin în etapele critice ale dezvoltării inimii fetale.
Factori Genetici
- Sindroame genetice: TAC este frecvent asociat cu anumite afecțiuni genetice. Cea mai cunoscută asociere este cu sindromul de deleție 22q11.2 (sindromul DiGeorge), prezent la aproximativ o treime din pacienți.
- Predispoziție ereditară: Existența unui părinte sau a unei rude de gradul I cu o malformație cardiacă congenitală poate crește riscul.
Factori de Mediu (Materni)
- Infecții virale: Anumite infecții contractate de mamă în timpul sarcinii, cum ar fi rubeola, pot afecta dezvoltarea cardiacă a fătului.
- Boli materne necontrolate: Diabetul zaharat sau fenilcetonuria maternă pot crește riscul.
- Expunerea la substanțe: Consumul de alcool, droguri sau anumite medicamente (retinoizi, unele anticonvulsivante) în timpul sarcinii sunt factori de risc cunoscuți.
🧪 Diagnosticare
Diagnosticul precis și rapid este crucial. Acesta poate fi stabilit atât înainte, cât și după naștere.
Diagnostic prenatal
Datorită progreselor în ecografia fetală, trunchiul arterial comun poate fi diagnosticat încă din timpul sarcinii, de obicei în trimestrul al doilea, în timpul ecografiei morfologice. O imagine anormală a cordului, în special în secțiunea “trei vase și trahee”, poate ridica suspiciunea. Un ecocardiogram fetal, realizat de un cardiolog pediatru, poate confirma diagnosticul, permițând părinților să primească consiliere și să planifice nașterea într-un centru specializat cu acces la chirurgie cardiacă pediatrică.
Diagnostic postnatal
După naștere, procesul de diagnosticare urmează mai mulți pași:
-
Examen Clinic și PulsoximetrieMedicul observă cianoza și ascultă suflul cardiac. Pulsoximetria (măsurarea saturației de oxigen) arată valori scăzute constant.
-
EcocardiografieAceasta este metoda de elecție pentru diagnostic. Vizualizează direct trunchiul unic, valva truncală, defectul septal ventricular și originea arterelor pulmonare.
-
Teste ComplementareElectrocardiograma (ECG) arată hipertrofie biventriculară (îngroșarea pereților ambilor ventriculi).
Radiografia toracică relevă cardiomegalie (inima mărătă) și semne de flux sanguin pulmonar crescut.
Cateterismul cardiac poate fi necesar pentru a clarifica anatomia coronariană și a măsura presiunile înainte de operație.
🧠 Verificarea cunoștințelor
În afară de trunchiul arterial unic, ce alt defect cardiac este aproape întotdeauna prezent în această malformație?
🩹 Tratament Chirurgical
Tratamentul pentru trunchiul arterial comun este exclusiv chirurgical și reprezintă o urgență. Fără intervenție, supraviețuirea este rară. Operația se realizează de obicei în primele săptămâni sau luni de viață.
Înainte de operație, se administrează un tratament medical pentru a stabiliza copilul și a controla simptomele de insuficiență cardiacă (cu diuretice și medicamente care ajută inima să pompeze mai eficient).
Obiectivele intervenției chirurgicale corective (operația Rastelli):
- Închiderea defectului septal ventricular (DSV): Se folosește un petic (patch) pentru a închide comunicarea dintre ventricule, astfel încât ventriculul stâng să pompeze sânge doar în trunchiul arterial comun.
- Crearea unei noi aorte: Trunchiul arterial comun original este lăsat pe loc pentru a funcționa ca noua aortă, care va transporta sângele oxigenat din ventriculul stâng către corp.
- Crearea unei noi artere pulmonare: Arterele pulmonare sunt deconectate de la trunchiul comun. Se creează o nouă cale de ieșire din ventriculul drept spre arterele pulmonare folosind un conduct (un tub sintetic cu sau fără valvă) sau un homogref (o valvă și o arteră umană donată).
Această operație complexă pe cord deschis restabilește o circulație sanguină normală, cu două circuite separate.
📈 Complicații și Prognostic
Prognosticul copiilor cu trunchi arterial comun s-a îmbunătățit dramatic datorită chirurgiei. Supraviețuirea postoperatorie este acum de peste 90% în centrele specializate. Cu toate acestea, pacienții necesită îngrijire cardiologică pe tot parcursul vieții.
Prognostic și provocări pe termen lung
> 90%
Foarte ridicată
Bună-Excelentă
Complicațiile pe termen lung pot include:
- Deteriorarea conductului: Conductul care leagă ventriculul drept de arterele pulmonare nu crește odată cu copilul și se poate îngusta (stenoză) sau uza. Majoritatea pacienților vor necesita una sau mai multe operații de înlocuire a conductului de-a lungul vieții.
- Disfuncția valvei truncale: Valva originală a trunchiului, care acum funcționează ca valvă aortică, poate prezenta scurgeri (regurgitare) sau îngustări (stenoză) și poate necesita reparație sau înlocuire.
- Aritmii: Pacienții pot dezvolta tulburări de ritm cardiac.
- Insuficiență cardiacă: Funcția ventriculelor poate fi afectată pe termen lung.
🛡️ Prevenție și Consiliere Genetică
Deoarece majoritatea cazurilor nu au o cauză clar identificabilă, prevenția este dificilă. Totuși, îngrijirea prenatală adecvată este importantă. Femeile însărcinate ar trebui să evite alcoolul, fumatul, drogurile și să discute cu medicul despre orice medicament pe care îl iau. Vaccinarea împotriva rubeolei înainte de sarcină și un bun control al bolilor cronice (cum ar fi diabetul) sunt măsuri esențiale.
Consiliere Genetică
Având în vedere asocierea puternică cu sindromul de deleție 22q11.2 și alte anomalii genetice, consilierea genetică este esențială pentru familiile afectate. Testarea genetică poate oferi un diagnostic complet, poate estima riscul de recurență în sarcinile viitoare și poate identifica alte posibile probleme de sănătate asociate sindromului.
❓ Întrebări Frecvente
Este o malformație cardiacă congenitală gravă în care un singur vas mare de sânge pleacă din inimă, în loc de două (aortă și arteră pulmonară), ducând la amestecul sângelui oxigenat cu cel neoxigenat și la simptome severe precum cianoza și insuficiența cardiacă.
Da, fără intervenție chirurgicală corectivă, prognosticul este extrem de sumbru. Majoritatea copiilor (aproximativ 85%) nu supraviețuiesc primului an de viață din cauza complicațiilor severe, în principal insuficiența cardiacă.
Da, mulți copii care au suferit o reparație chirurgicală reușită pot duce vieți active și împlinite. Totuși, este crucială monitorizarea cardiologică pe tot parcursul vieții, deoarece vor fi necesare probabil intervenții chirurgicale sau intervenționale ulterioare pentru a înlocui conductul pulmonar. Pot exista anumite restricții privind activitățile fizice foarte intense.
Frecvența reintervențiilor depinde de tipul de conduct utilizat și de ritmul de creștere al copilului. Conducturile se uzează și devin prea mici în timp. De obicei, prima înlocuire este necesară în copilărie sau adolescență. Progresele medicale permit astăzi înlocuirea unor conducte prin proceduri minim invazive (cateterism cardiac), evitând operațiile pe cord deschis.
📚 Resurse Suplimentare
Informațiile prezentate în acest articol sunt consolidate din surse medicale de încredere. Pentru o înțelegere mai aprofundată a anatomiei generale și a contextului cardiovascular, puteți consulta următoarele resurse (în limba engleză):
Anatomy, Head and Neck: Carotid Arteries – StatPearls – NCBI
URL: www.ncbi.nlm.nih.gov/books/NBK545238/
Common carotid artery
URL: en.wikipedia.org/wiki/Common_carotid_artery
Common carotid artery: Anatomy
URL: www.kenhub.com/en/library/anatomy/common-carotid-artery
What Are The Carotid Arteries?
URL: my.clevelandclinic.org/health/body/21492-carotid-artery
Carotid artery: Anatomy, function, disease, and more
URL: www.medicalnewstoday.com/articles/carotid-artery
Major Arteries of the Head and Neck – Carotid
URL: teachmeanatomy.info/neck/vessels/arterial-supply/











