
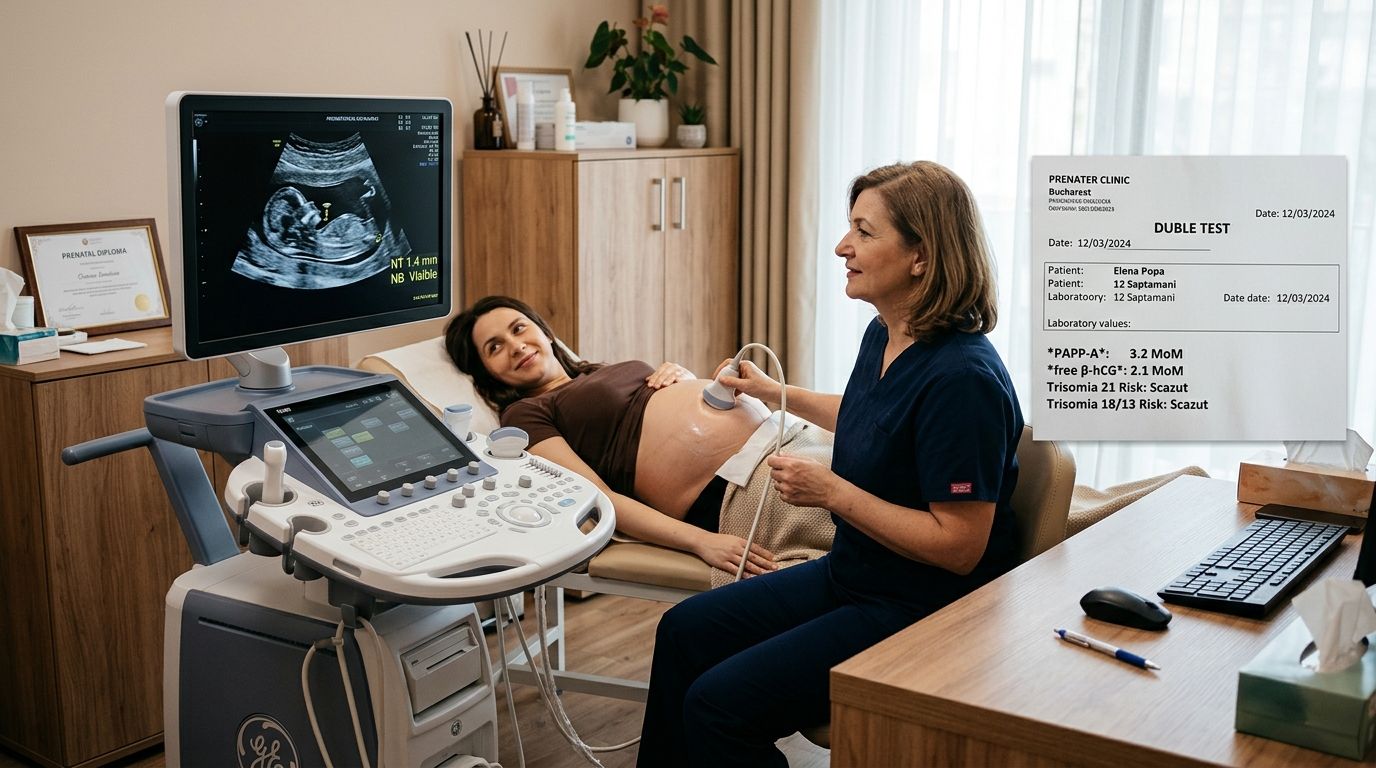
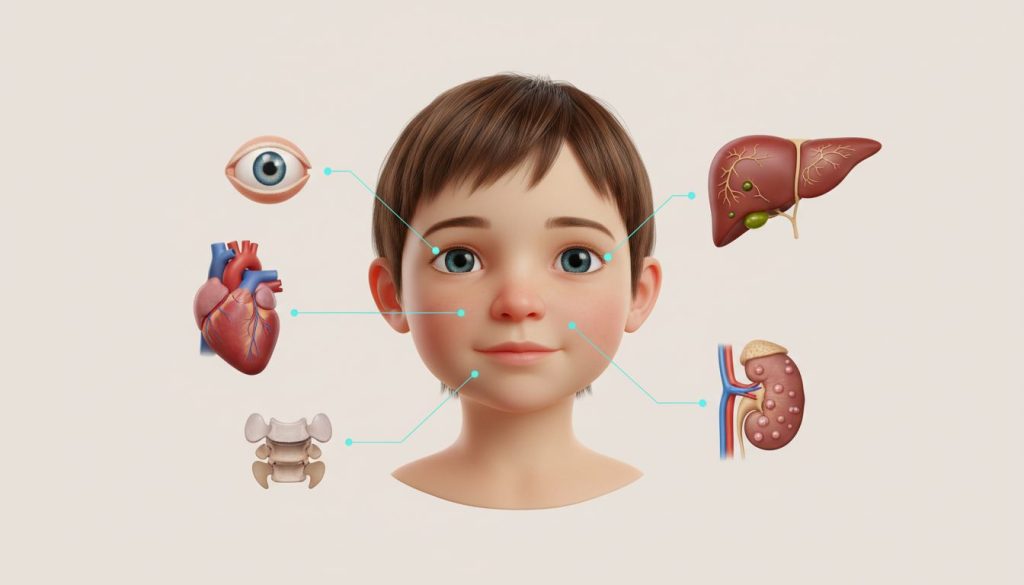
Sindromul Alagille (ALGS) este o tulburare genetică complexă și rară care afectează multiple organe, cel mai adesea ficatul, inima, ochii, scheletul și rinichii. Caracterizată în principal prin colestază cronică (un flux biliar redus sau blocat de la ficat), afecțiunea este cauzată de un număr insuficient de canale biliare intrahepatice. Această acumulare de bilă în ficat duce la leziuni progresive, prurit sever (mâncărime intensă), icter și poate evolua spre ciroză. Pe lângă afectarea hepatică, pacienții prezintă adesea malformații cardiace congenitale, un aspect facial distinctiv, anomalii vertebrale și oculare.
Deoarece nu există un tratament care să vindece sindromul, managementul este multidisciplinar și se concentrează pe ameliorarea simptomelor și prevenirea complicațiilor. Tratamentul include medicamente pentru a îmbunătăți fluxul biliar și a controla pruritul, suplimente de vitamine, o dietă bogată în calorii și, în cazuri severe de boală hepatică, transplantul de ficat, care poate schimba dramatic prognosticul. Supraviețuirea pe termen lung depinde în mare măsură de severitatea afectării cardiace și hepatice.
- 🧬 Cauză Genetică: O mutație în gena JAG1 (majoritatea cazurilor) sau NOTCH2, transmisă autosomal dominant sau apărută spontan (de novo).
- 💔 Afectare Multisistemică: Implică în principal ficatul (colestază), inima (ex. Tetralogia Fallot), scheletul (vertebre “în fluture”), ochii și fața (facies tipic).
- 🔍 Diagnostic Bazat pe Criterii: Necesită prezența a cel puțin 3 din 5 criterii majore: colestază, anomalii cardiace, scheletice, oculare și facies caracteristic.
- 💊 Management Simptomatic: Nu există vindecare; tratamentul vizează controlul pruritului, susținerea nutriției și corectarea defectelor cardiace. Transplantul hepatic este o opțiune în cazuri grave.
- 👶 Debut Precoce: Simptomele, în special icterul, apar de obicei în primele luni de viață, necesitând o diagnosticare rapidă pentru un management adecvat.
Cuprins
🧬 Ce este Sindromul Alagille?
Sindromul Alagille (ALGS), cunoscut și sub denumirea de displazie arteriohepatică, este o afecțiune genetică rară, cu transmitere autosomal dominantă, care poate afecta numeroase sisteme și organe. Prevalența sa este estimată la 1 din 30.000 până la 1 din 50.000 de nașteri vii, afectând în mod egal ambele sexe și toate grupurile etnice.
La baza sindromului se află mutații ale unor gene esențiale pentru dezvoltarea embrionară, în special a căilor de semnalizare Notch. Acestea duc la o dezvoltare anormală a mai multor structuri, dar cea mai distinctivă trăsătură este paucity of intrahepatic bile ducts (un număr redus de canale biliare mici în interiorul ficatului). Aceste canale sunt vitale pentru transportul bilei, un lichid produs de ficat pentru a ajuta la digestia grăsimilor. Cu mai puține canale, bila se acumulează în ficat, o condiție numită colestază, provocând leziuni hepatice progresive.
Mutațiile genetice responsabile sunt localizate în principal pe gena JAG1 (prezentă la 70-90% dintre pacienți) și, mai rar, pe gena NOTCH2. Deși este o boală genetică, aproximativ 30-50% dintre cazuri apar spontan (mutații de novo), la copii fără istoric familial. Simptomatologia debutează de obicei în primele trei luni de viață și poate varia dramatic în severitate, chiar și între membrii aceleiași familii. Unii indivizi pot avea o formă atât de ușoară încât rămâne nediagnosticată, în timp ce alții se confruntă cu complicații severe, care le pun viața în pericol și pot necesita transplant hepatic sau intervenții cardiace complexe.
🌡️ Simptomele sindromului Alagille
Spectrul clinic al sindromului Alagille este extrem de larg. Simptomele pot fi prezente de la naștere sau se pot dezvolta în copilărie. Severitatea variază considerabil de la un pacient la altul.
Simptome comune
Cele mai frecvente manifestări sunt legate de cele cinci sisteme principale afectate:
- Afectare hepatică (Colestază cronică): Este piatra de temelie a diagnosticului.
- Icter persistent: Îngălbenirea pielii și a albului ochilor, care apare la nou-născut și persistă peste primele săptămâni de viață.
- Prurit sever: Mâncărime intensă și cronică pe tot corpul, cauzată de acumularea de acizi biliari în piele. Este adesea cel mai debilitant simptom, afectând somnul și calitatea vieții.
- Urină închisă la culoare (colurică) și scaune decolorate (aholice): Semne ale blocajului biliar.
- Hepatosplenomegalie: Mărirea ficatului și a splinei, detectabilă la palparea abdomenului.
- Xantome: Depozite gălbui de colesterol sub piele, în special pe coate, genunchi, fese și în jurul ochilor, cauzate de hipercolesterolemie.
- Deficit de creștere și malabsorbție: Din cauza digestiei deficitare a grăsimilor și a vitaminelor liposolubile (A, D, E, K), mulți copii au dificultăți în a lua în greutate.
- Anomalii cardiovasculare: Prezente la peste 90% dintre pacienți.
- Stenoza arterei pulmonare periferice: Cea mai frecventă anomalie, constând în îngustarea ramurilor arterei pulmonare.
- Tetralogia Fallot: O combinație complexă de patru defecte cardiace, prezentă la aproximativ 7-13% dintre pacienți. Poate cauza cianoză (colorație albăstruie a pielii), dispnee (dificultăți de respirație) și fatigabilitate.
- Alte defecte includ șant septal atrial sau ventricular și canal arterial patent.
- Facies tipic: Mulți pacienți (dar nu toți) au trăsături faciale distinctive, care devin mai evidente cu vârsta:
- Frunte lată și proeminentă.
- Ochi adânciți și distanțați (hipertelorism).
- Nas drept cu o bază largă și vârf bulbat.
- Bărbie mică și ascuțită (micrognație), care dă feței un aspect triunghiular.
- Anomalii scheletice:
- Vertebre “în fluture” (sau “în scrumieră”): O anomalie a arcurilor vertebrale care nu fuzionează corect, vizibilă pe radiografiile toracice. De obicei, nu cauzează probleme medicale, dar este un indiciu diagnostic important (prezent la 50-70% dintre pacienți).
- Anomalii oculare:
- Embriotoxon posterior: O linie opacă, proeminentă, pe suprafața internă a corneei. Este prezent la peste 80% dintre pacienți și este de obicei detectabil doar la un examen oftalmologic specializat. Nu afectează vederea.
Pacienți cu anomalii cardiace
Cazuri cauzate de mutația JAG1
Pacienți cu embriotoxon posterior
Necesită transplant hepatic
Simptome rare
Pe lângă manifestările clasice, pot apărea și alte probleme:
- Afectare renală: Pot exista diverse anomalii, de la chisturi renale la acidoză tubulară renală, care pot progresa spre insuficiență renală.
- Hipertensiune portală: Creșterea presiunii în vena portă, ca o complicație a bolii hepatice cronice, putând duce la varice esofagiene.
- Anomalii vasculare: Unii pacienți pot dezvolta anevrisme sau disecții ale vaselor de sânge, inclusiv la nivelul creierului (sindromul Moyamoya), reprezentând un risc serios de accident vascular cerebral.
- Întârzieri în dezvoltare: Pot apărea întârzieri în dezvoltarea motorie sau cognitivă, parțial din cauza malnutriției cronice și a spitalizărilor frecvente.
🧬 Cauze și factori de risc
Sindromul Alagille este o boală monogenică, ceea ce înseamnă că este cauzată de o mutație într-o singură genă. Principalii vinovați sunt:
- Gena JAG1: Localizată pe cromozomul 20, mutațiile în această genă sunt responsabile pentru 70-90% din cazuri.
- Gena NOTCH2: Localizată pe cromozomul 1, mutații mai rare în această genă cauzează restul cazurilor.
Ambele gene fac parte din calea de semnalizare “Notch”, un sistem de comunicare intercelulară crucial în timpul dezvoltării embrionare pentru formarea corectă a multor organe, inclusiv ficatul, inima, vasele de sânge și scheletul.
Moștenire și Risc
Principalul factor de risc este prezența unui istoric familial de sindrom Alagille. Datorită variabilității extreme (expresivitate variabilă), un părinte poate avea o formă foarte ușoară, nediagnosticată, și poate avea un copil cu o formă severă a bolii. Diagnosticul prenatal este posibil prin testare genetică (amniocenteză sau prelevarea de vilozități coriale), dar este efectuat de obicei doar în familiile cu un caz cunoscut.
🧠 Verifică-ți cunoștințele
Care este cea mai frecventă mutație genetică asociată cu Sindromul Alagille?
🩺 Diagnostic
Diagnosticarea sindromului Alagille este un proces complex, care implică o combinație de evaluare clinică, teste de laborator, imagistică și, în final, confirmare genetică.
Metode de diagnostic
Procesul de diagnosticare urmează, de regulă, mai mulți pași:
-
Suspiciune clinicăMedicul suspectează ALGS pe baza simptomelor, cum ar fi icterul neonatal prelungit, un suflu cardiac sau trăsăturile faciale caracteristice.
-
Investigații inițialeSe efectuează analize de sânge (pentru funcția hepatică), ecografie abdominală (pentru a evalua ficatul și căile biliare), ecocardiogramă (pentru inimă) și examen oftalmologic.
-
Criterii de diagnostic & BiopsieDiagnosticul clasic se bazează pe prezența a cel puțin 3 din 5 criterii majore. O biopsie hepatică este adesea crucială pentru a demonstra numărul redus de ducte biliare (raport ducte/spații porte < 0.5).
-
Confirmare GeneticăTestarea genetică prin secvențiere poate identifica mutația în gena JAG1 sau NOTCH2, confirmând diagnosticul în 90-95% din cazurile cu suspiciune clinică puternică.
Criteriile clinice majore pentru diagnostic sunt:
- Colestază cronică (confirmată prin biopsie hepatică ce arată o lipsă de ducte biliare).
- Anomalii cardiovasculare (cel mai adesea stenoza arterei pulmonare).
- Anomalii scheletice (vertebre “în fluture”).
- Anomalii oculare (embriotoxon posterior).
- Facies caracteristic.
Prezența a cel puțin trei dintre aceste cinci caracteristici stabilește diagnosticul clinic.
Când să consulți un medic
Este esențial ca părinții să solicite o evaluare medicală dacă observă următoarele semne la un nou-născut sau copil mic:
- Icter (piele sau ochi galbeni) care durează mai mult de 2 săptămâni.
- Mâncărime severă a pielii, fără o cauză aparentă (erupție cutanată).
- Creștere lentă în greutate și înălțime.
- Detectarea unui suflu cardiac de către medicul pediatru la un control de rutină.
- Scaune foarte deschise la culoare (albe sau gri) și urină închisă la culoare.
💊 Tratament și Management
Nu există un tratament curativ pentru sindromul Alagille. Managementul este unul de susținere, multidisciplinar, și se concentrează pe tratarea simptomelor specifice fiecărui pacient și pe prevenirea complicațiilor. Echipa medicală include de obicei un hepatolog pediatru, un cardiolog, un oftalmolog, un nefrolog și un nutriționist.
Managementul colestazei și al pruritului
- Acid ursodeoxicolic (Ursodiol): Medicament de primă linie care ajută la îmbunătățirea fluxului biliar și poate reduce pruritul și nivelul colesterolului.
- Medicamente pentru prurit: Atunci când Ursodiolul nu este suficient, se pot folosi colestiramină (leagă acizii biliari în intestin), rifampicină, naltrexonă sau antihistaminice.
- Suplimente de vitamine: Este crucială administrarea de doze mari de vitamine liposolubile (A, D, E, K) pentru a preveni deficiențele cauzate de malabsorbție.
Managementul nutrițional
- Dietă bogată în calorii: Copiii cu ALGS au nevoi energetice crescute.
- Trigliceride cu lanț mediu (MCT): Se folosesc formule speciale sau uleiuri MCT, deoarece acestea sunt mai ușor de absorbit în absența unui flux biliar normal.
Managementul cardiac și chirurgical
- Corecție chirurgicală cardiacă: Multe defecte cardiace, cum ar fi Tetralogia Fallot, necesită intervenții chirurgicale corective în copilărie.
- Derivație biliară parțială externă (PEBD): O procedură chirurgicală în care o mică secțiune a intestinului este adusă la suprafața pielii pentru a drena o parte din bilă în afara corpului, într-o pungă. Poate reduce dramatic pruritul și xantomele la pacienții rezistenți la tratament medicamentos.
- Transplant hepatic: Este opțiunea finală pentru pacienții (15-25%) care dezvoltă boală hepatică în stadiu terminal (ciroză, hipertensiune portală necontrolată) sau au prurit intratabil care distruge calitatea vieții.
Avantajele Transplantului Hepatic
- Singura opțiune curativă pentru boala hepatică.
- Îmbunătățește dramatic calitatea vieții (dispariția pruritului, a icterului).
- Corectează anomaliile metabolice (hipercolesterolemia).
- Rate de supraviețuire post-transplant de 80-90% la 5 ani.
Riscuri și Dezavantaje
- Necesită imunosupresie pe viață, cu riscuri de infecții și cancer.
- Complicații chirurgicale (sângerare, tromboză vasculară).
- Riscul cardiac perioperator este crescut din cauza anomaliilor preexistente.
- Nu rezolvă problemele extrahepatice (cardiace, renale, vasculare).
⚠️ Complicații
Progresia bolii poate duce la o serie de complicații grave, care necesită monitorizare atentă:
- Ciroză și insuficiență hepatică: Aproximativ 20% dintre pacienți vor dezvolta ciroză, necesitând în cele din urmă un transplant.
- Hipertensiune portală: Poate duce la splenomegalie (mărirea splinei), ascită (lichid în abdomen) și varice esofagiene (cu risc de sângerare masivă).
- Malnutriție severă și deficit de creștere: O provocare constantă în copilărie.
- Fracturi patologice: Din cauza deficitului de vitamina D și a osteoporozei.
- Complicații vasculare: Anevrisme intracraniene sau în alte artere, cu risc de ruptură și hemoragie.
- Afectare cognitivă și dificultăți de învățare: Pot fi legate de malnutriție cronică, episoade de boală sau impactul psihosocial.
Mortalitatea pe termen lung este de 10-20% în absența unui transplant și este cel mai adesea legată de complicațiile cardiace sau boala hepatică terminală.
❤️ Stil de viață și Prevenție
Deși nu există o metodă de a preveni apariția sindromului Alagille, un management proactiv al stilului de viață poate îmbunătăți semnificativ starea de sănătate și calitatea vieții.
Sfaturi practice pentru pacienți și familii
Managementul pruritului: Păstrați pielea bine hidratată cu creme emoliente. Mențineți unghiile tăiate scurt pentru a preveni leziunile prin scărpinat. Băile cu apă călduță (nu fierbinte) pot oferi o ameliorare temporară.
Activitate fizică: Discutați cu medicul despre activitățile fizice sigure. Sporturile de contact ar trebui evitate de către pacienții cu splenomegalie, din cauza riscului de ruptură splenică.
Consiliere genetică: Familiile afectate ar trebui să beneficieze de consiliere genetică pentru a înțelege riscurile de transmitere și opțiunile de testare prenatală.
Prevenția primară nu este posibilă, deoarece este o tulburare genetică. Prevenția secundară se concentrează pe monitorizarea atentă pentru depistarea precoce a complicațiilor (ecografii anuale, endoscopii, evaluări cardiace) și pe managementul riguros al dietei și medicației.
🚻 Diferențe de tratament (Sex/Vârstă)
Sindromul Alagille afectează în mod egal bărbații și femeile, iar abordarea terapeutică nu diferă în funcție de sex. Totuși, prioritățile managementului se schimbă odată cu vârsta:
- La sugari și copii: Accentul principal este pus pe diagnosticarea rapidă, managementul colestazei și, cel mai important, pe asigurarea unei nutriții adecvate pentru a susține creșterea și dezvoltarea. Intervențiile chirurgicale cardiace corective sunt, de asemenea, o prioritate în această perioadă.
- La adolescenți și adulți: Pe măsură ce pacientul crește, focusul se mută spre monitorizarea și managementul complicațiilor pe termen lung, cum ar fi progresia bolii hepatice spre ciroză, hipertensiunea portală, sănătatea osoasă și monitorizarea anomaliilor vasculare. Tranziția de la îngrijirea pediatrică la cea pentru adulți este un pas critic care necesită o planificare atentă.
❓ Întrebări frecvente
▼
Gravitatea variază enorm. Unii oameni au simptome ușoare și duc o viață relativ normală, în timp ce alții dezvoltă complicații severe, care necesită transplant hepatic sau intervenții cardiace. Este o boală progresivă, dar cu un management medical adecvat, aproximativ 50-75% dintre pacienți supraviețuiesc până la vârsta adultă (>20 de ani).
▼
Nu. Este o boală strict genetică, cauzată de o mutație. Nu poate fi transmisă de la o persoană la alta prin contact.
▼
Prognosticul depinde în principal de severitatea afectării cardiace și hepatice. Problemele cardiace sunt principala cauză de deces în primul an de viață. Pentru cei care supraviețuiesc copilăriei, evoluția bolii hepatice devine factorul determinant. Transplantul hepatic poate schimba dramatic traiectoria bolii, oferind o speranță de viață mult mai lungă și o calitate a vieții net superioară.
📚 Referințe / Surse
Alagille’s Syndrome Symptoms in Children – UPMC Children’s Hospital. www.chp.edu/our-services/transplant/liver/education/liver-disease-states/alagilles-syndrome
Alagille Syndrome – Symptoms, Causes, Treatment | NORD. rarediseases.org/rare-diseases/alagille-syndrome/
Treatment for Alagille Syndrome – NIDDK.NIH.gov. www.niddk.nih.gov/health-information/liver-disease/alagille-syndrome/treatment
What Is Alagille Syndrome? – Cleveland Clinic. my.clevelandclinic.org/health/diseases/23540-alagille-syndrome
Alagille Syndrome | Liver Canada. liver.ca/alagille-syndrome/
Alagille Syndrome | Boston Children’s Hospital. www.childrenshospital.org/conditions-treatments/alagille-syndrome






